

Mechanistically driven development of iridium catalysts for asymmetric allylic substitution
- PMID: 20873839
- PMCID: PMC3008324
- DOI: 10.1021/ar100047x
Mechanistically driven development of iridium catalysts for asymmetric allylic substitution
Abstract
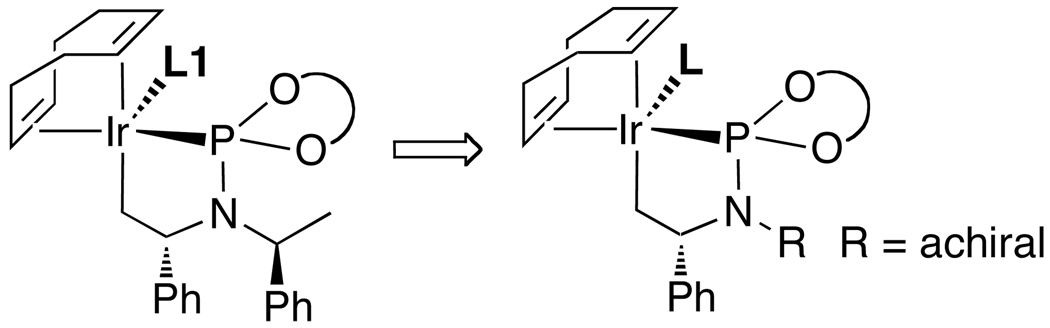
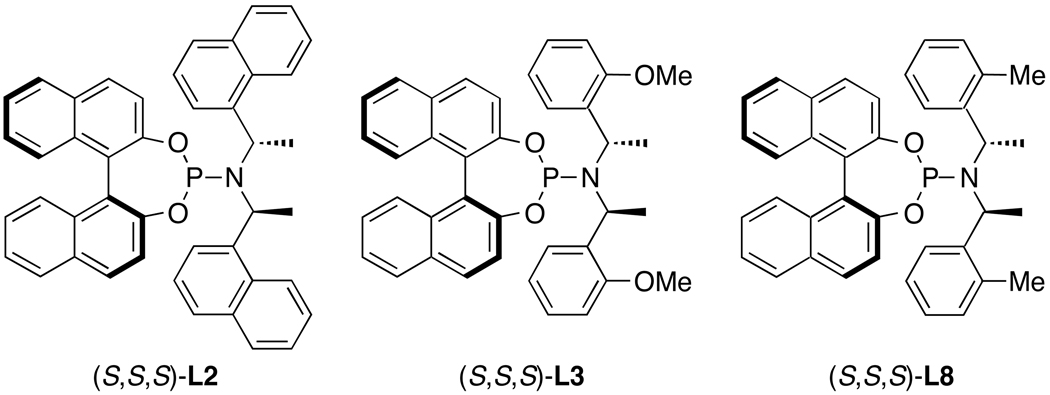
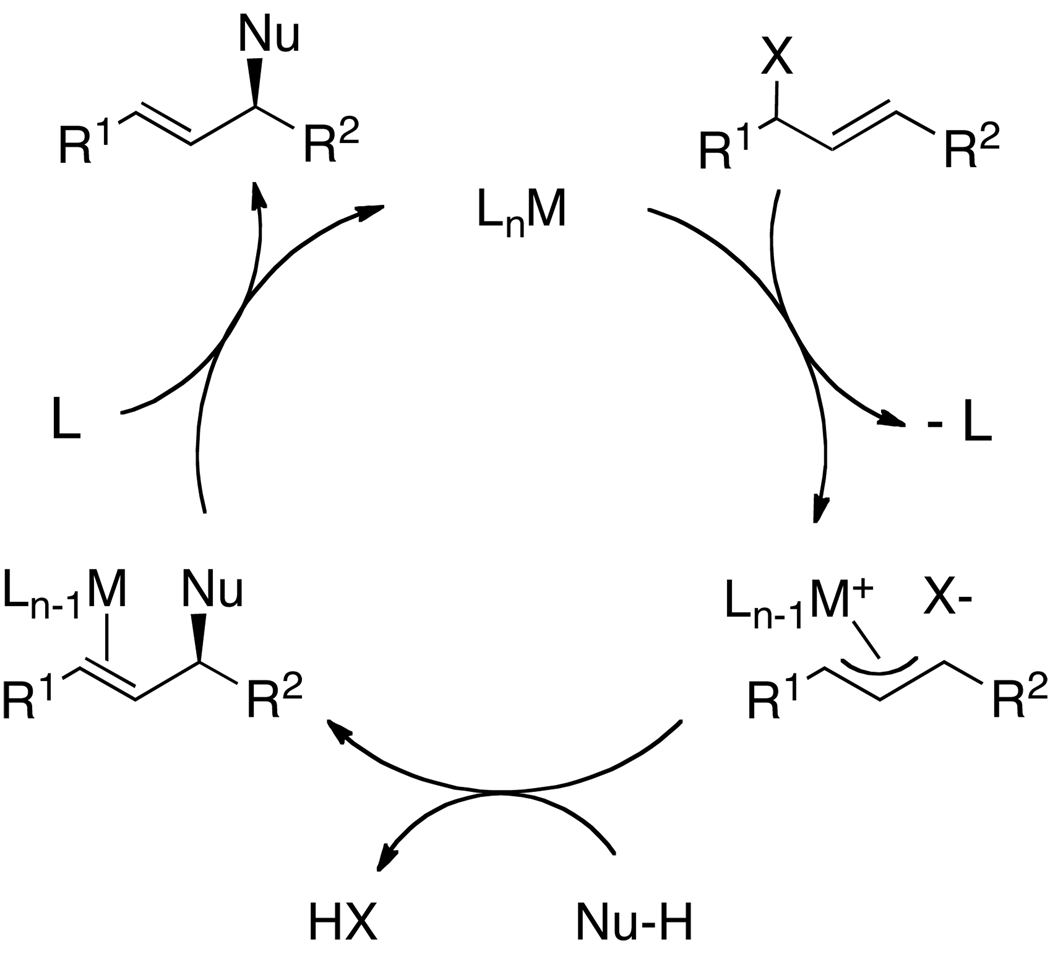
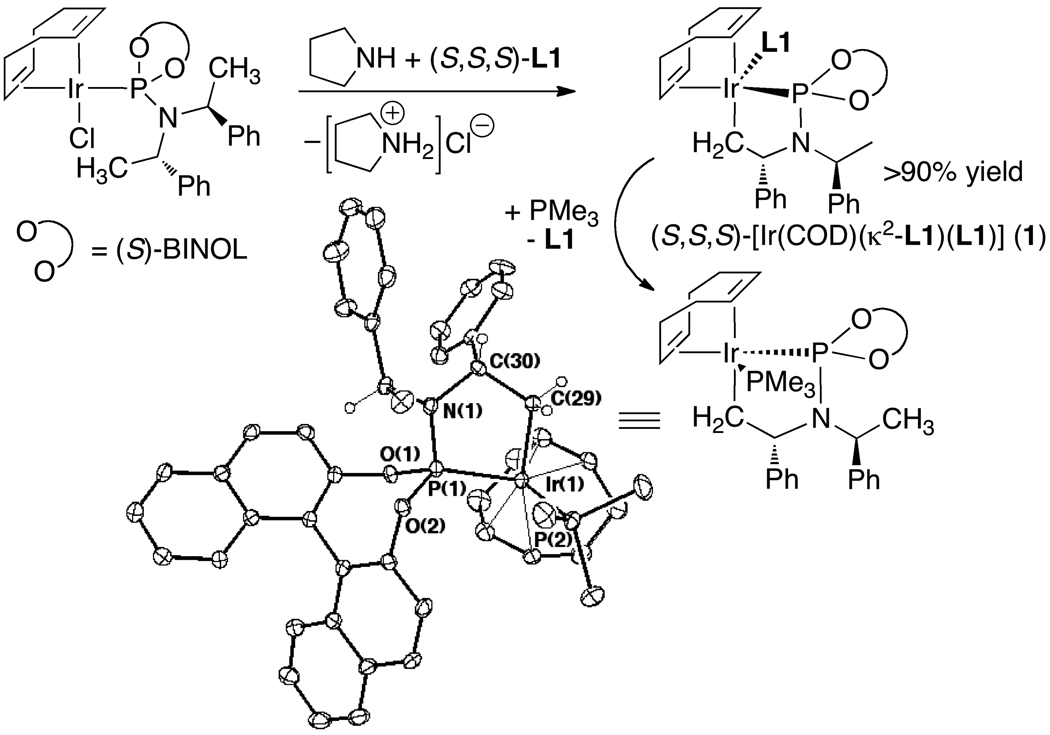
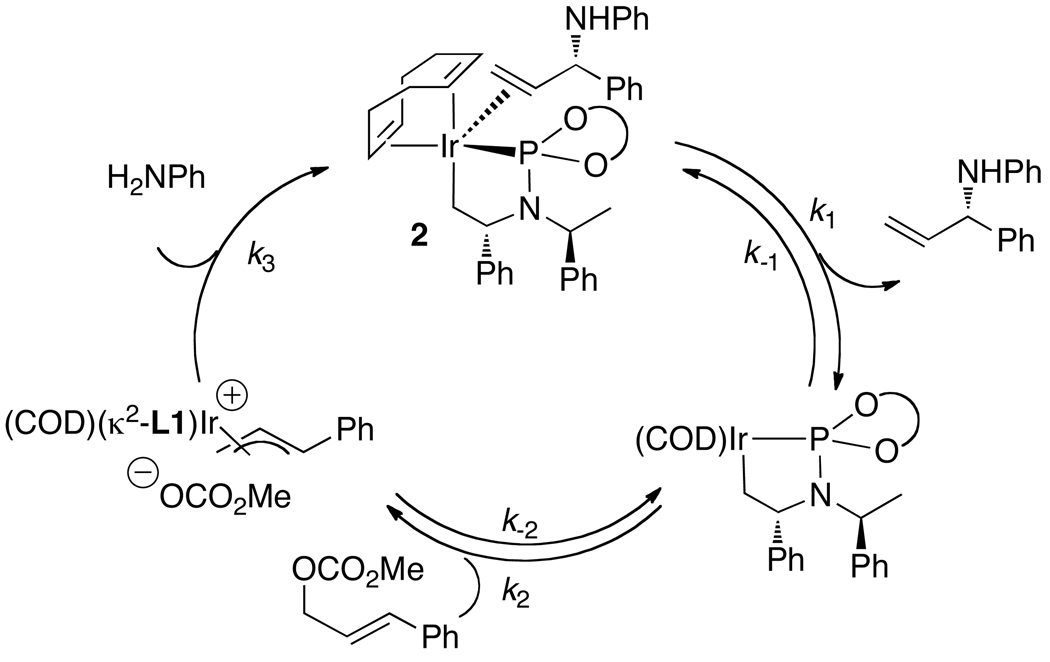
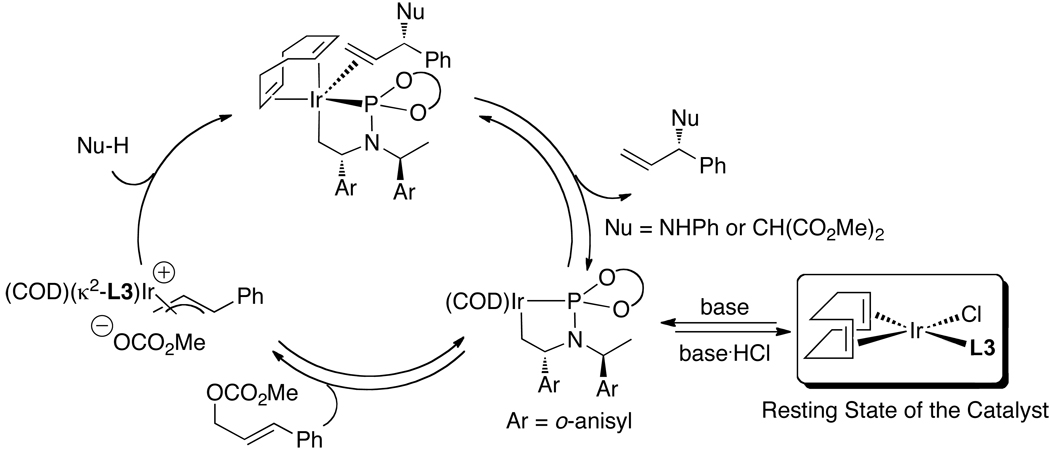
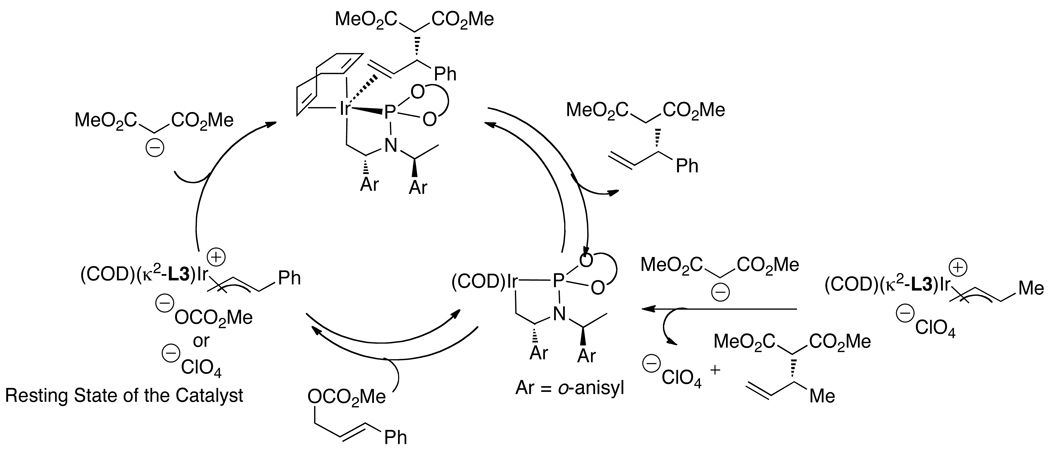
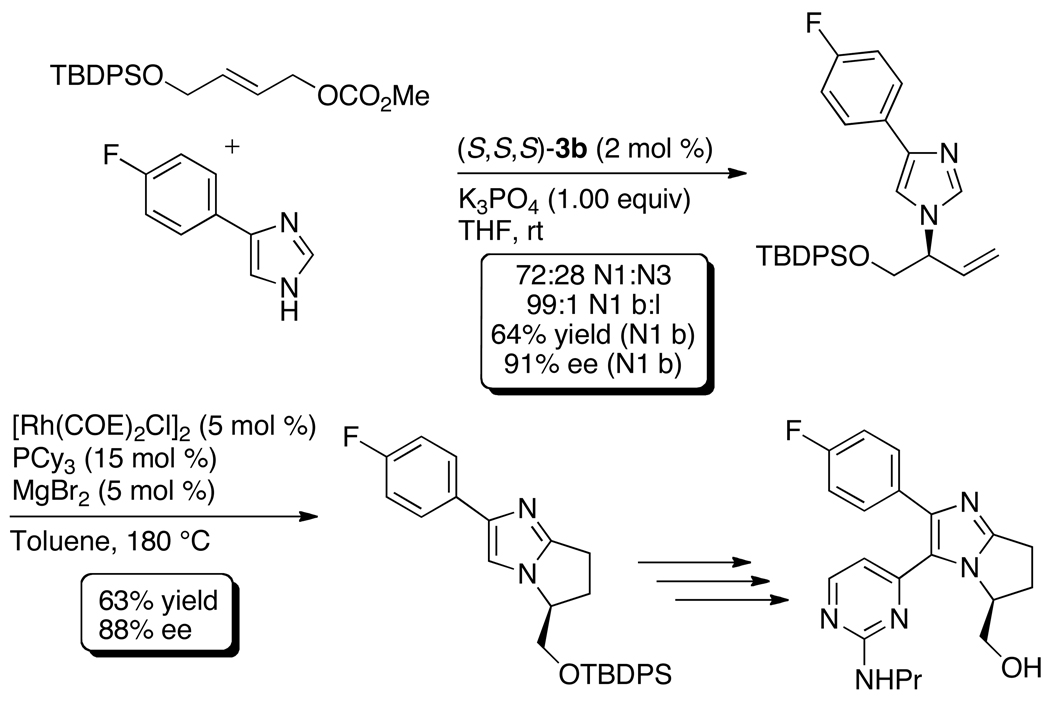
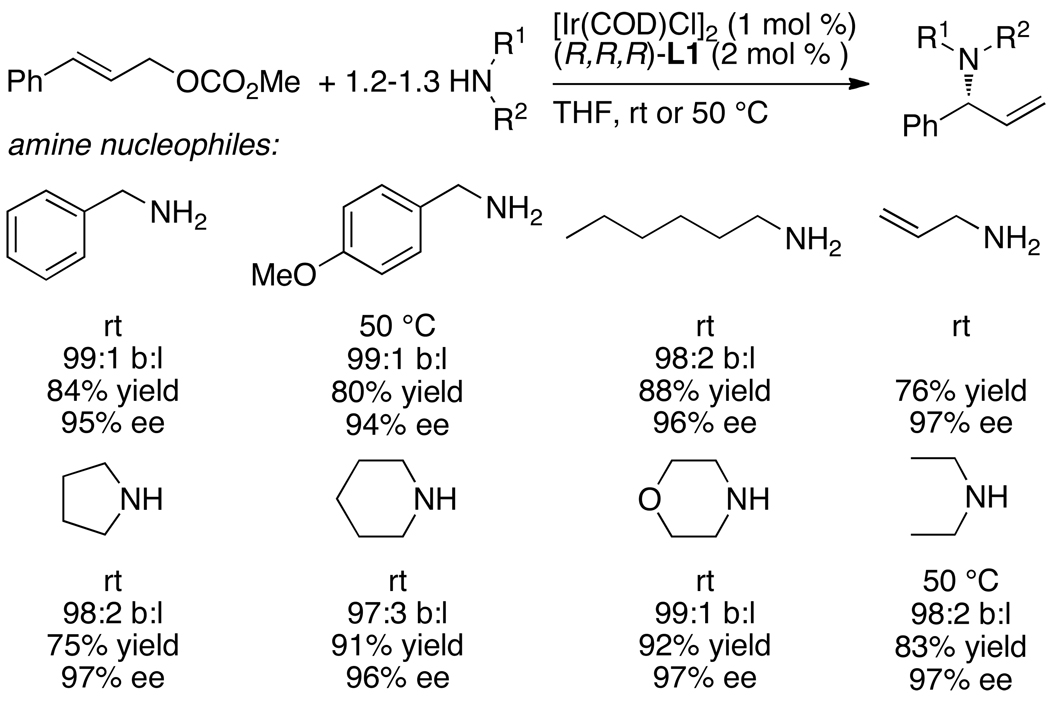
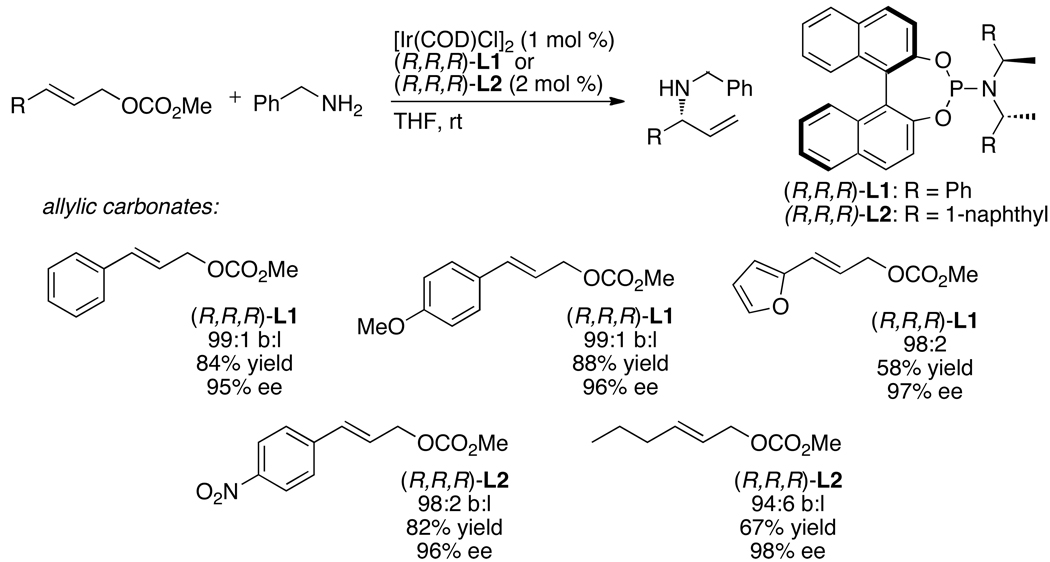
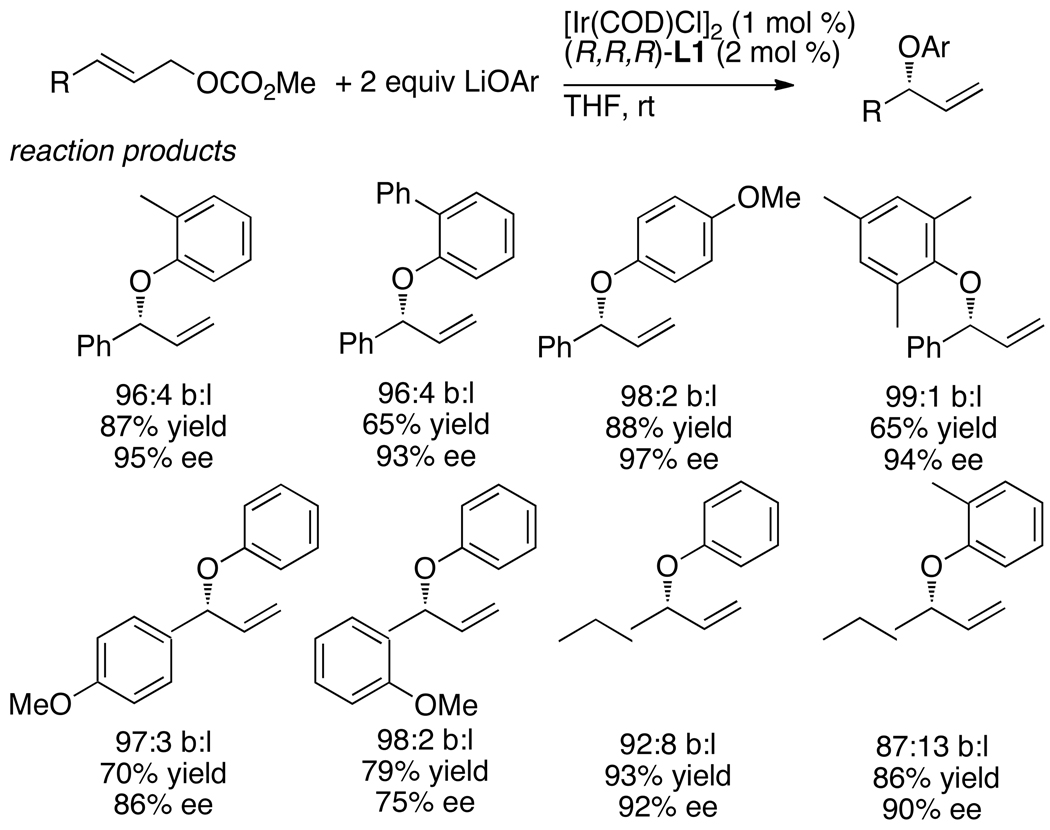
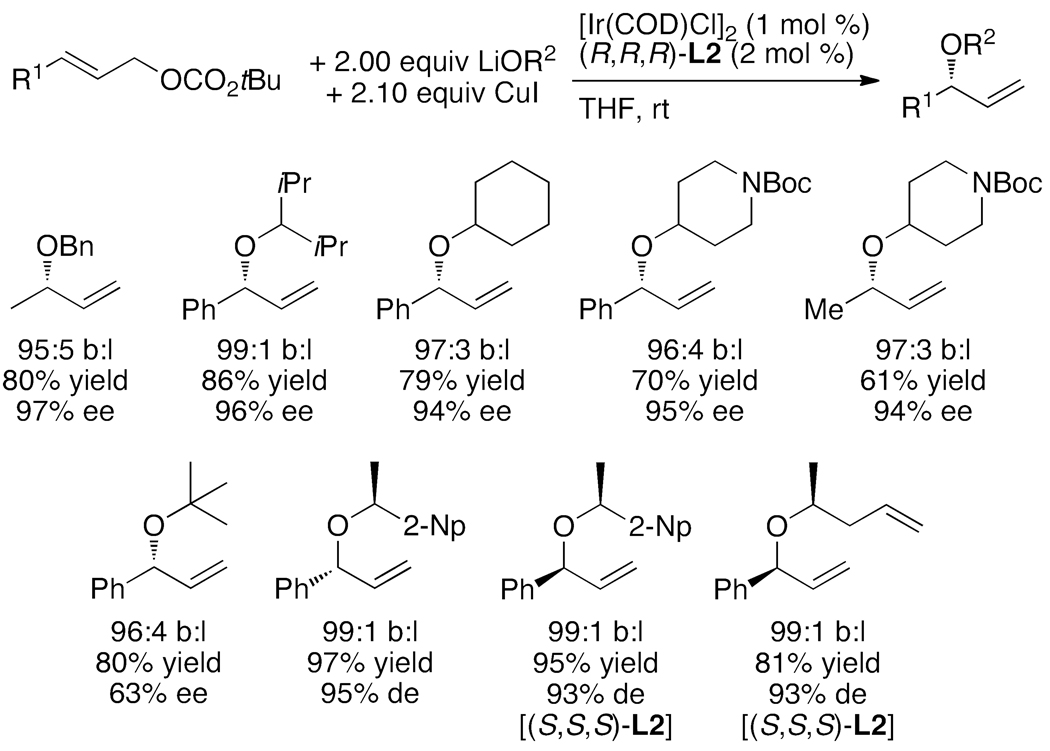
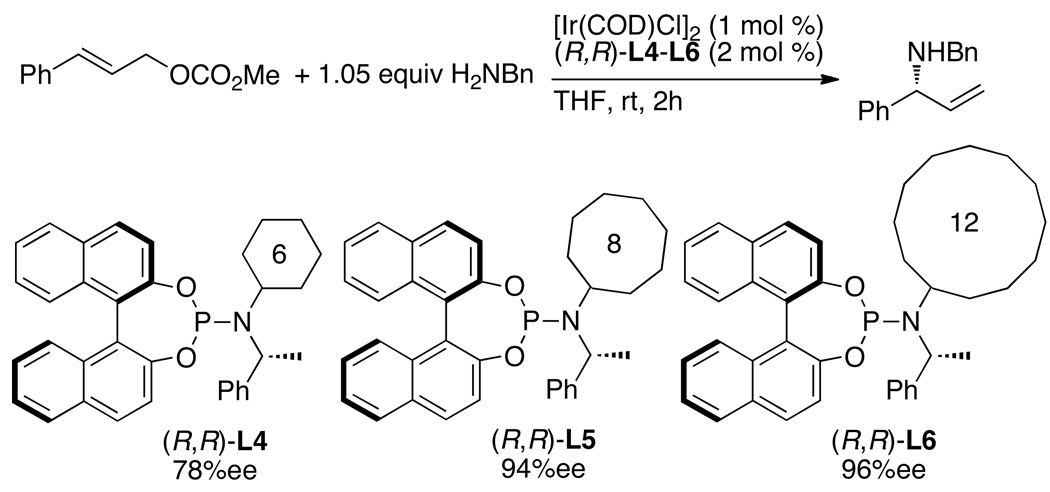
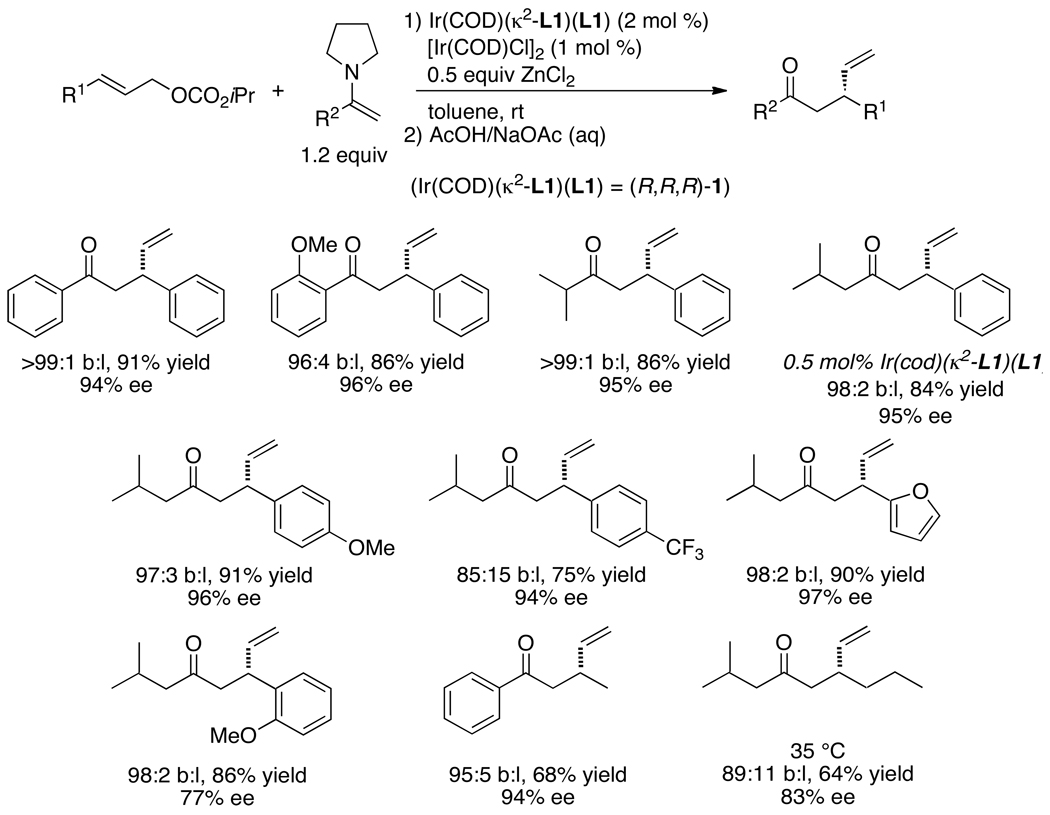
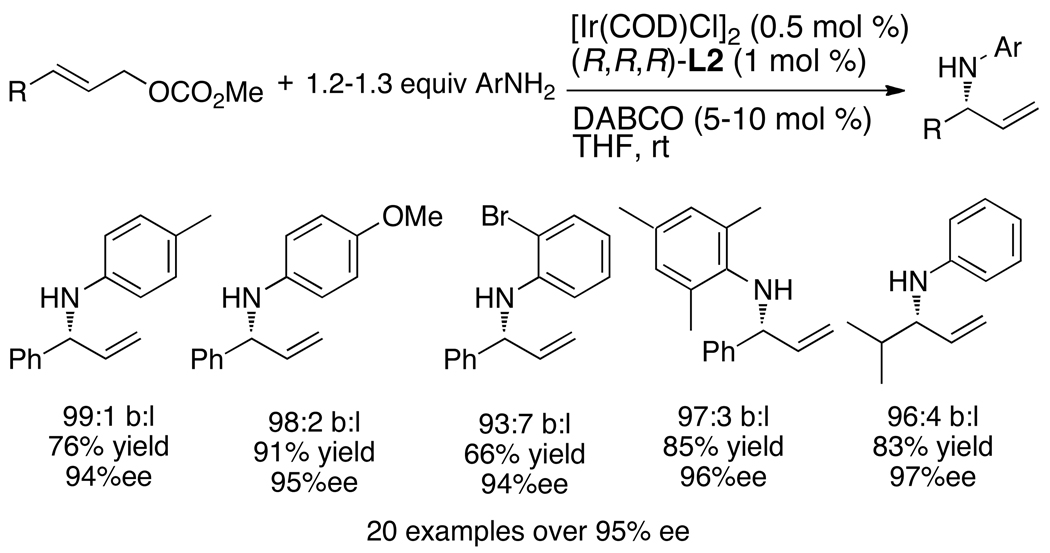
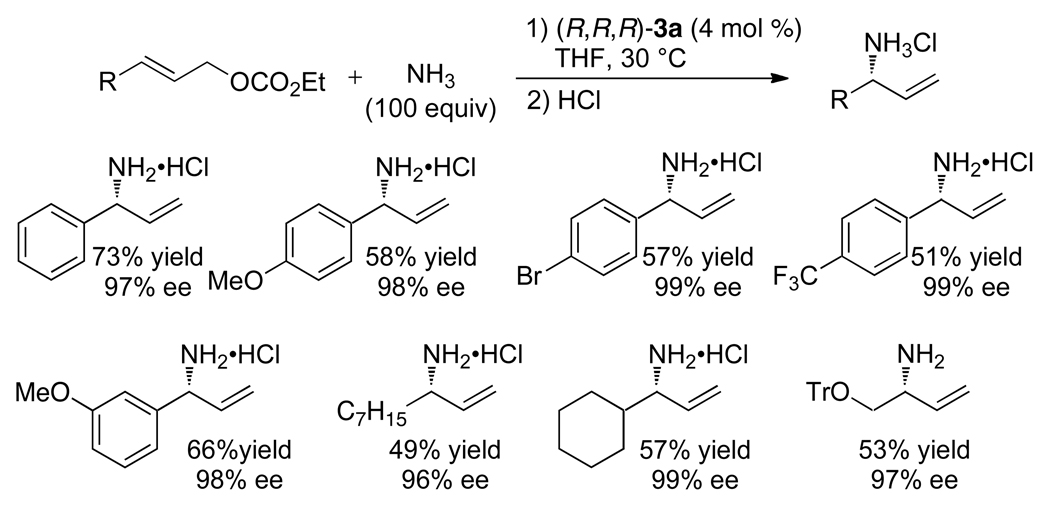
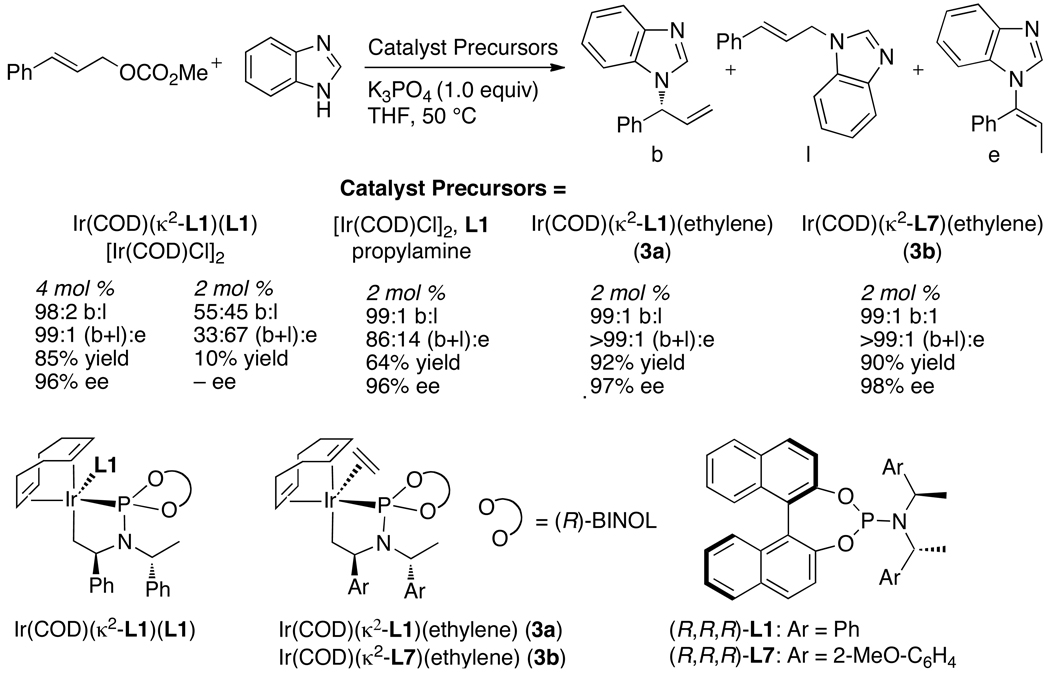
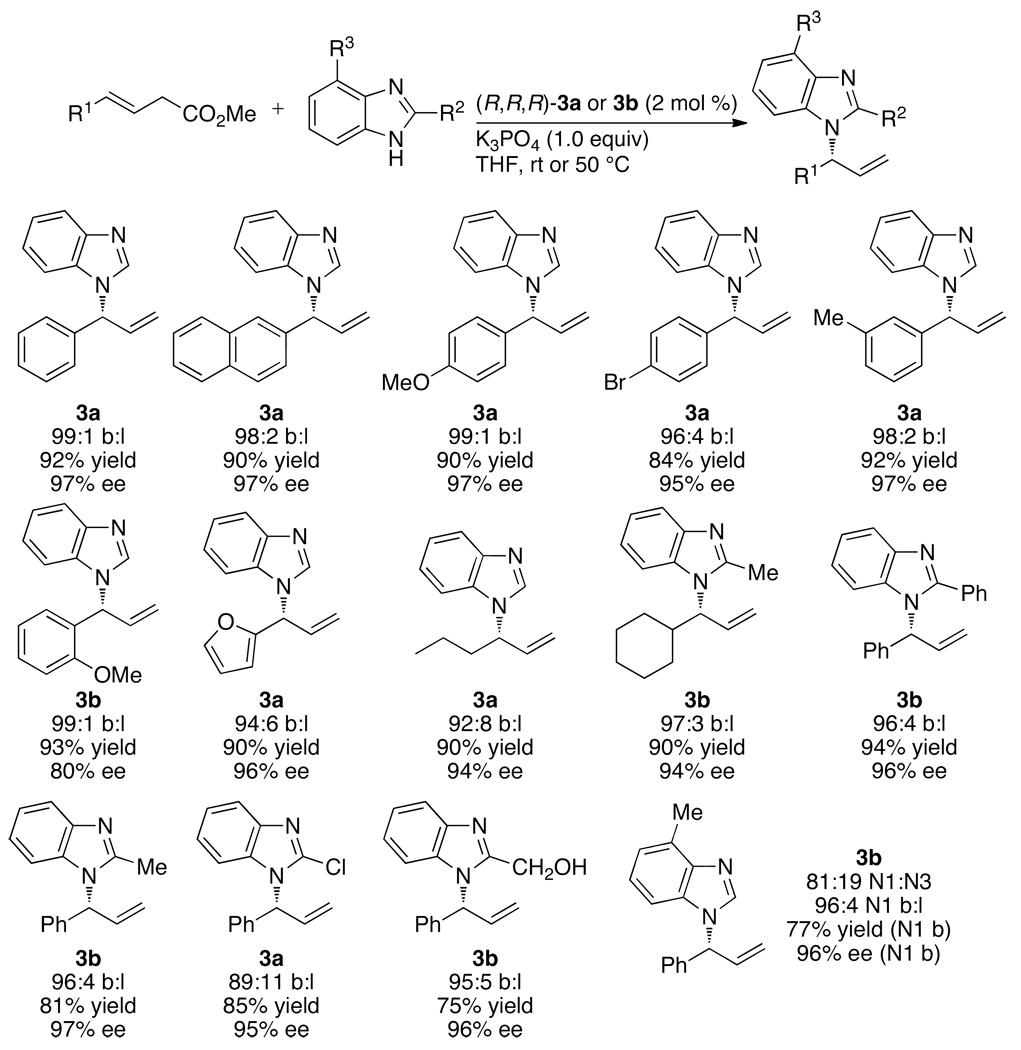
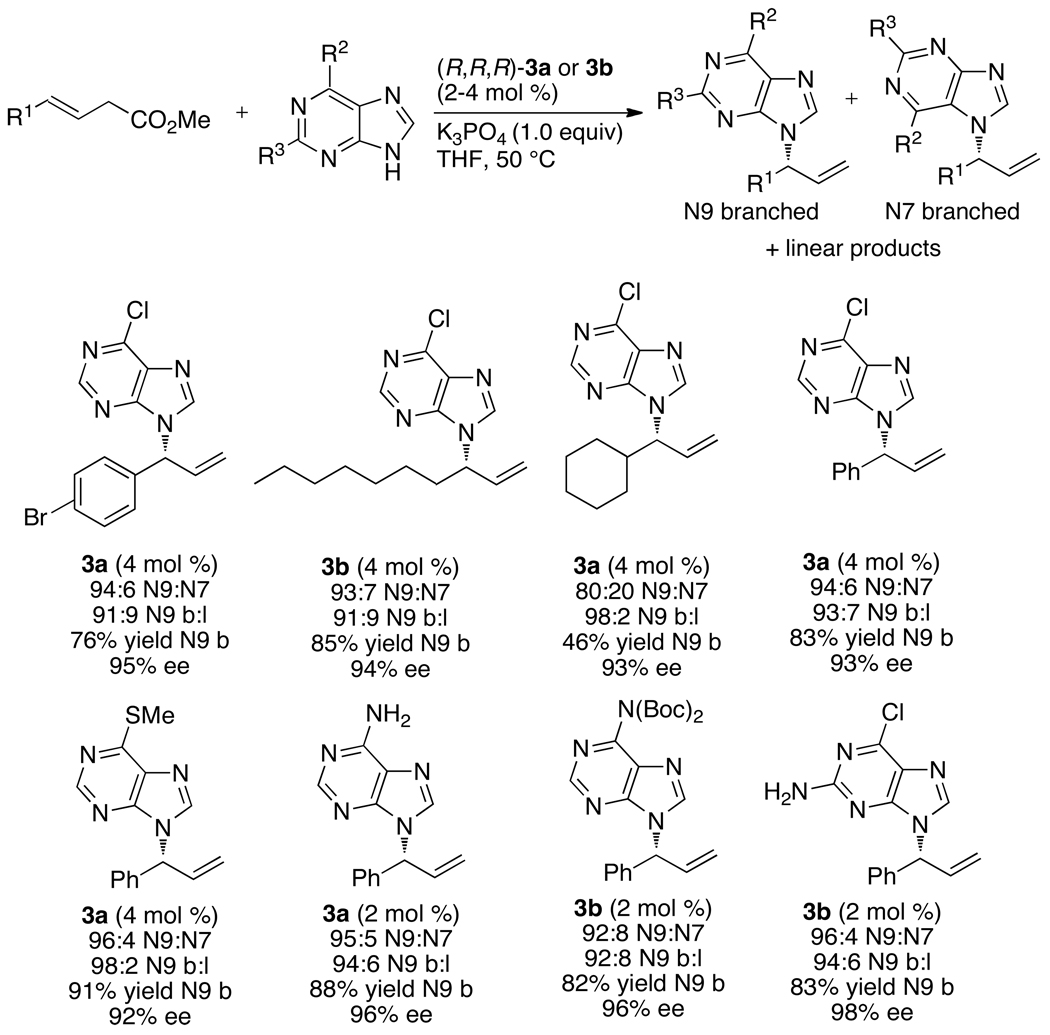
Enantioselective allylic substitution reactions comprise some of the most versatile methods for preparing enantiomerically enriched materials. These reactions form products that contain multiple functionalities by creating carbon-nitrogen, carbon-oxygen, carbon-carbon, and carbon-sulfur bonds. For many years, the development of catalysts for allylic substitution focused on palladium complexes. However, studies of complexes of other metals have revealed selectivities that often complement those of palladium systems. Most striking is the observation that reactions with unsymmetrical allylic electrophiles that typically occur with palladium catalysts at the less hindered site of an allylic electrophile occur at the more hindered site with catalysts based on other metals. In this Account, we describe the combination of an iridium precursor and a phosphoramidite ligand that catalyzes enantioselective allylic substitution reactions with a particularly broad scope of nucleophiles. The active form of this iridium catalyst is not generated by the simple binding of the phosphoramidite ligand to the metal precursor. Instead, the initial phosphoramidite and iridium precursor react in the presence of base to form a metallacyclic species that is the active catalyst. This species is generated either in situ or separately in isolated form by reactions with added base. The identification of the structure of the active catalyst led to the development of simplified catalysts as well as the most active form of the catalyst now available, which is stabilized by a loosely bound ethylene. Most recently, this structure was used to prepare intermediates containing allyl ligands, the structures of which provide a model for the enantioselectivities discussed here. Initial studies from our laboratory on the scope of iridium-catalyzed allylic substitution showed that reactions of primary and secondary amines, including alkylamines, benzylamines, and allylamines, and reactions of phenoxides and alkoxides occurred in high yields, with high branched-to-linear ratios and high enantioselectivities. Parallel mechanistic studies had revealed the metallacyclic structure of the active catalyst, and subsequent experiments with the purposefully formed metallacycle increased the reaction scope dramatically. Aromatic amines, azoles, ammonia, and amides and carbamates as ammonia equivalents all reacted with high selectivities and yields. Moreover, weakly basic enolates (such as silyl enol ethers) and enolate equivalents (such as enamines) also reacted, and other research groups have used this catalyst to conduct reactions of stabilized carbon nucleophiles in the absence of additional base. One hallmark of the reactions catalyzed by this iridium system is the invariably high enantioselectivity, which reflects a high stereoselectivity for formation of the allyl intermediate. Enantioselectivity typically exceeds 95%, regioselectivity for formation of branched over linear products is usually near 20:1, and yields generally exceed 75% and are often greater than 90%. Thus, the development of iridium catalysts for enantioselective allylic substitution shows how studies of reaction mechanism can lead to a particularly active and a remarkably general system for an enantioselective process. In this case, a readily accessible catalyst effects allylic substitution, with high enantioselectivity and regioselectivity complementary to that of the venerable palladium systems.
Figures
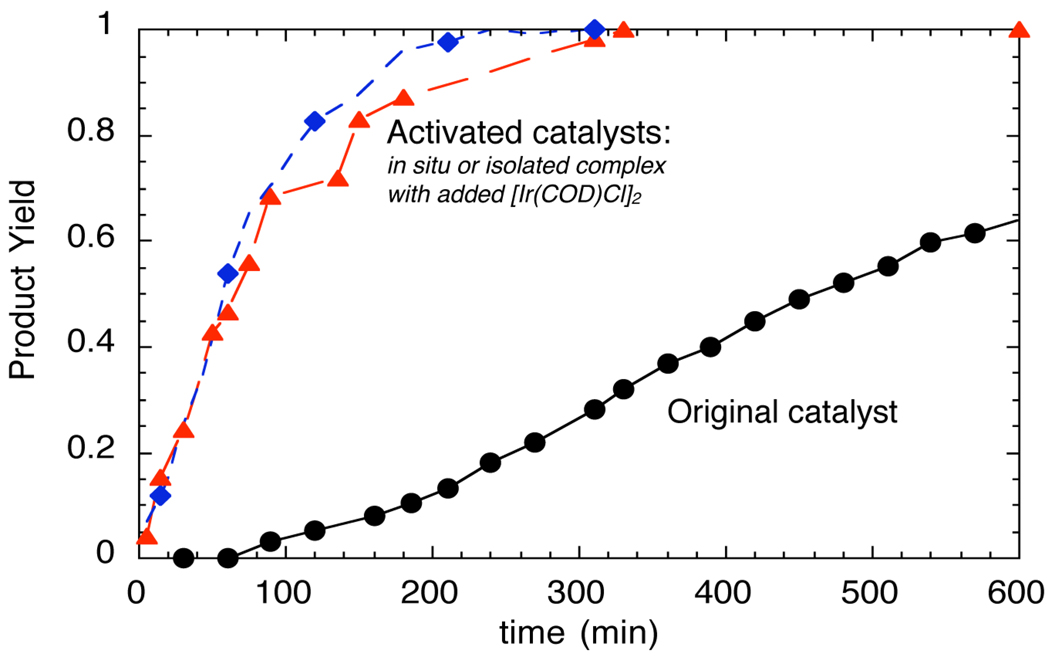
 ), the combination of isolated metallacyclic complex 1 and [Ir(COD)Cl]2 (red,
), the combination of isolated metallacyclic complex 1 and [Ir(COD)Cl]2 (red,  ), or the original system generated by combining [Ir(COD)Cl]2 and L1 (black, ●).
), or the original system generated by combining [Ir(COD)Cl]2 and L1 (black, ●).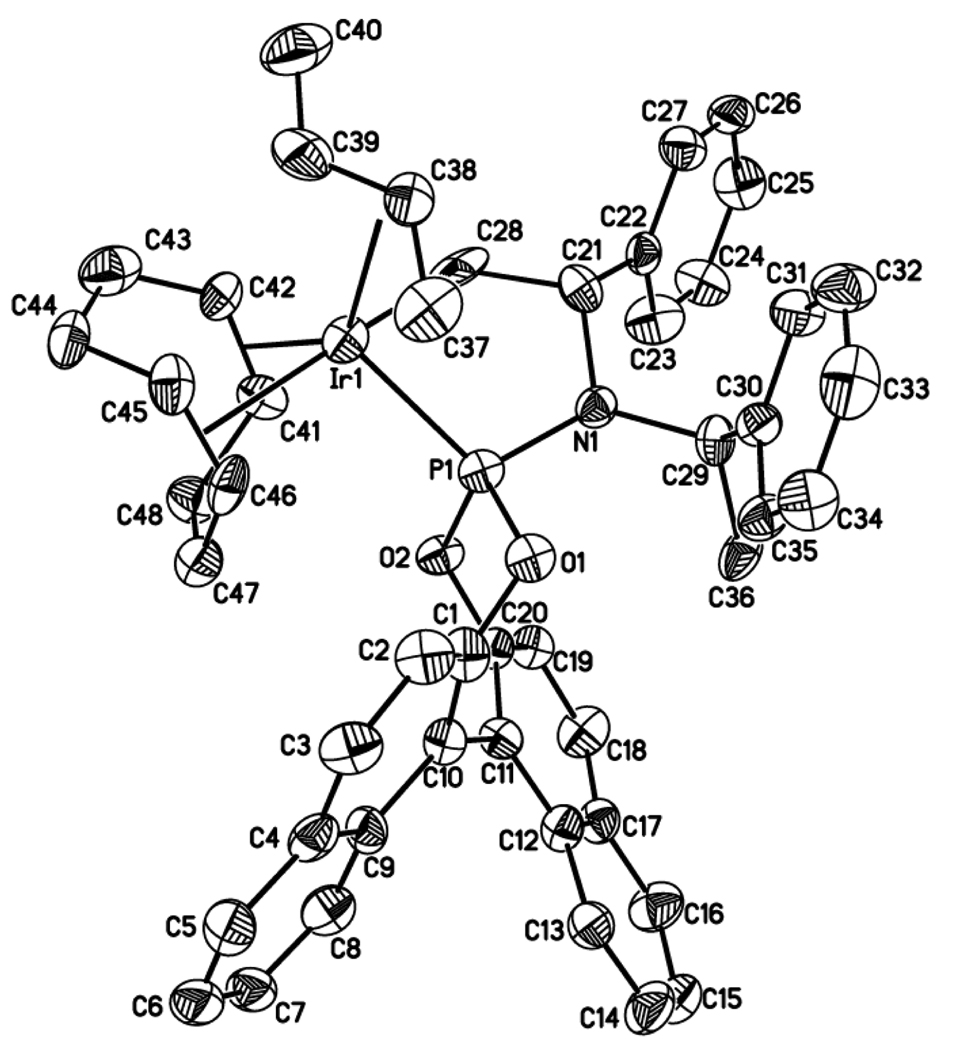

References
-
- Hartwig JF. Organotransition Metal Chemistry. Sausalito, CA: University Science Books; 2010. Chapter 20: Allylic Substitution; pp. 967–1014.
-
- Lu Z, Ma SM. Metal-Catalyzed Enantioselective Allylation in Asymmetric Synthesis. Angew. Chem. Int. Ed. 2008;47:258–297. - PubMed
-
- Trost BM. Pd Asymmetric Allylic Alkylation (AAA). A Powerful Synthetic Tool. Chem. Pharm. Bull. 2002;50:1–14. - PubMed
-
- Trost BM. Asymmetric Allylic Alkylation, an Enabling Methodology. J. Org. Chem. 2004;69:5813–5837. - PubMed
-
-
For a recent system and a discussion of prior work by Hayashi and Pfaltz, see You SL, Zhu XZ, Luo YM, Hou XL, Dai LX. Highly Regio-and Enantioselective Pd-Catalyzed Allylic Alkylation and Amination of Monosubstituted Allylic Acetates with Novel Ferrocene P,N-Ligands. J. Am. Chem. Soc. 2001;123:7471–7472.
-
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
