

Genome-wide model for the normal eukaryotic DNA replication fork
- PMID: 20876092
- PMCID: PMC2955150
- DOI: 10.1073/pnas.1010178107
Genome-wide model for the normal eukaryotic DNA replication fork
Abstract
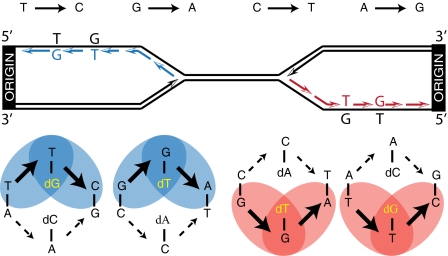
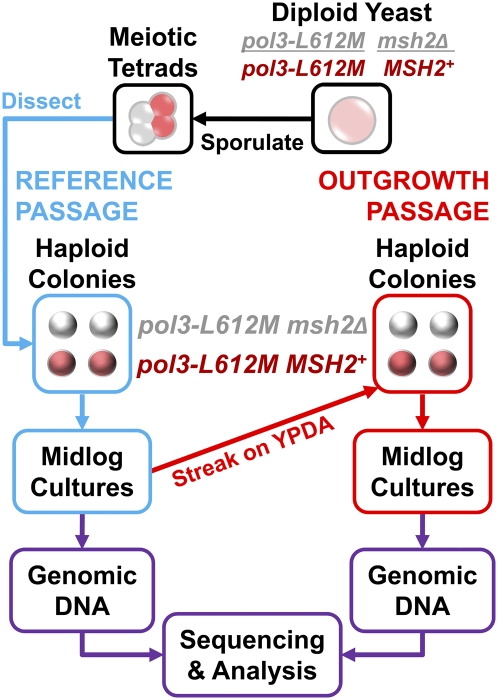
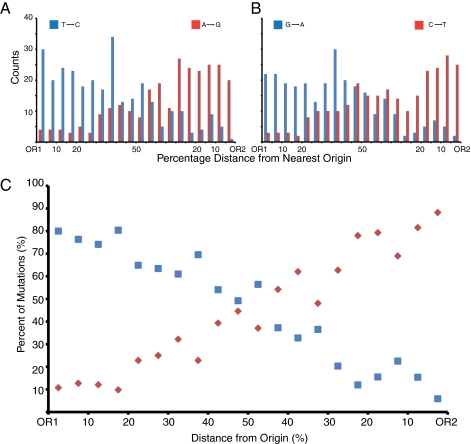
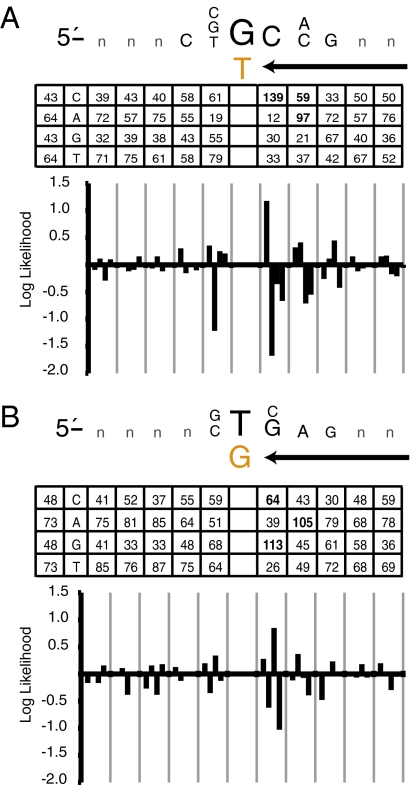
To investigate DNA replication enzymology across the nuclear genome of budding yeast, deep sequencing was used to establish the pattern of uncorrected replication errors generated by an asymmetric mutator variant of DNA polymerase δ (Pol δ). Sequencing of 16 genomes identified 1,206-bp substitutions generated over 33 generations by L612M Pol δ in a mismatch repair defective strain. Alignment of sequences flanking these substitutions identified "hotspot" motifs for Pol δ replication errors. The substitutions were distributed evenly across all 16 chromosomes. The vast majority were transitions that occurred with a strand bias that varied in a predictable manner relative to known functional origins of replication. This strand bias strongly supports the idea that Pol δ is primarily a lagging strand polymerase during replication across the entire nuclear genome.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
