

Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes
- PMID: 20876107
- PMCID: PMC2955112
- DOI: 10.1073/pnas.1004487107
Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes
Abstract
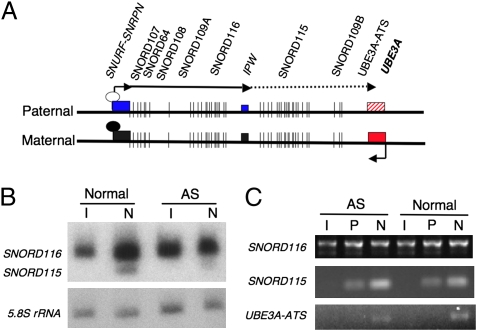
Angelman syndrome (AS) and Prader-Willi syndrome (PWS) are neurodevelopmental disorders of genomic imprinting. AS results from loss of function of the ubiquitin protein ligase E3A (UBE3A) gene, whereas the genetic defect in PWS is unknown. Although induced pluripotent stem cells (iPSCs) provide invaluable models of human disease, nuclear reprogramming could limit the usefulness of iPSCs from patients who have AS and PWS should the genomic imprint marks be disturbed by the epigenetic reprogramming process. Our iPSCs derived from patients with AS and PWS show no evidence of DNA methylation imprint erasure at the cis-acting PSW imprinting center. Importantly, we find that, as in normal brain, imprinting of UBE3A is established during neuronal differentiation of AS iPSCs, with the paternal UBE3A allele repressed concomitant with up-regulation of the UBE3A antisense transcript. These iPSC models of genomic imprinting disorders will facilitate investigation of the AS and PWS disease processes and allow study of the developmental timing and mechanism of UBE3A repression in human neurons.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
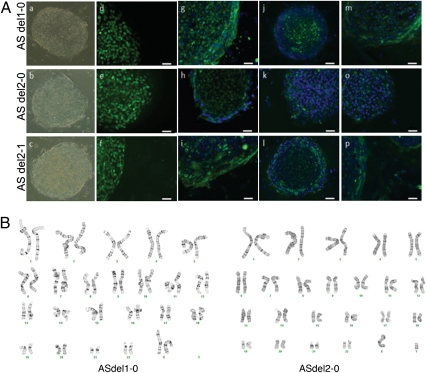
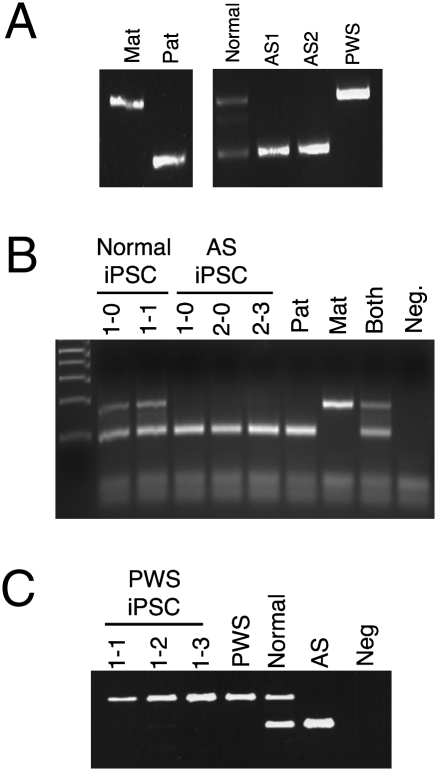
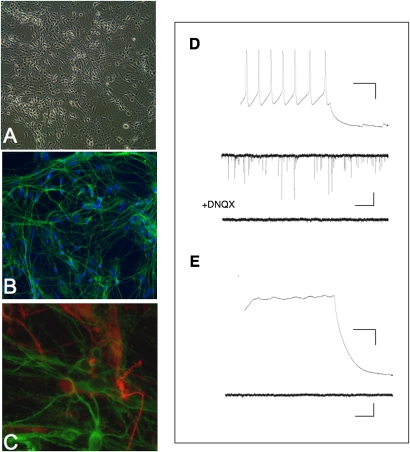
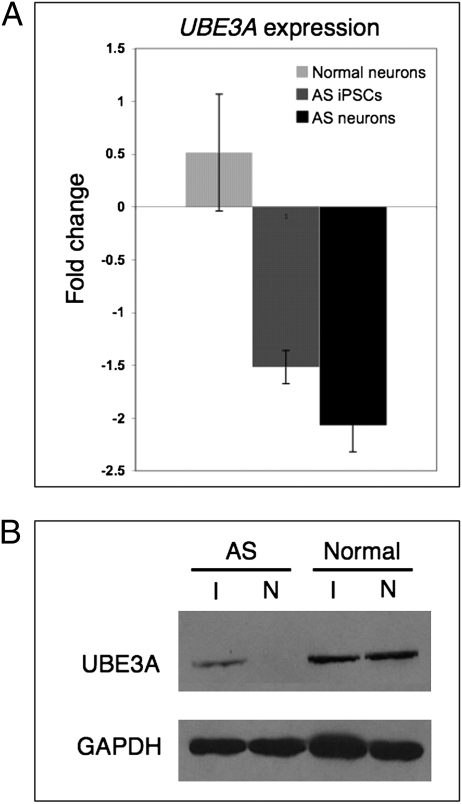
References
-
- Williams CA, et al. Angelman syndrome 2005: Updated consensus for diagnostic criteria. Am J Med Genet A. 2006;140:413–418. - PubMed
-
- Rougeulle C, Glatt H, Lalande M. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat Genet. 1997;17:14–15. - PubMed
-
- Vu TH, Hoffman AR. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat Genet. 1997;17:12–13. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
