

Oncogenic HER2{Delta}16 suppresses miR-15a/16 and deregulates BCL-2 to promote endocrine resistance of breast tumors
- PMID: 20876285
- PMCID: PMC2994280
- DOI: 10.1093/carcin/bgq192
Oncogenic HER2{Delta}16 suppresses miR-15a/16 and deregulates BCL-2 to promote endocrine resistance of breast tumors
Abstract
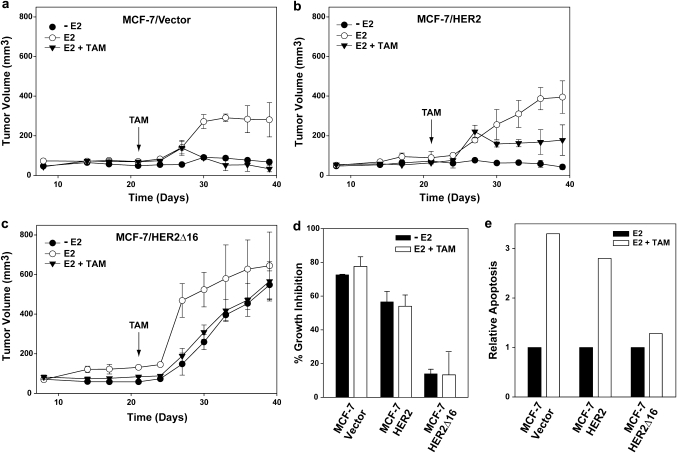
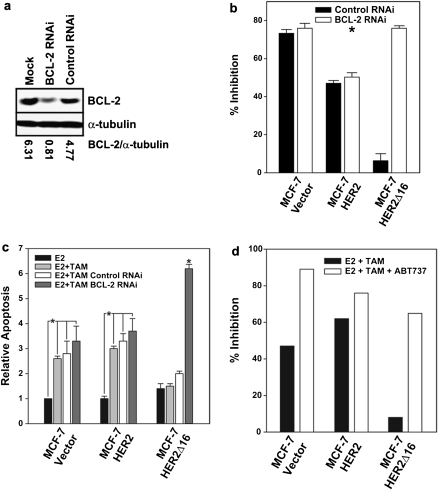
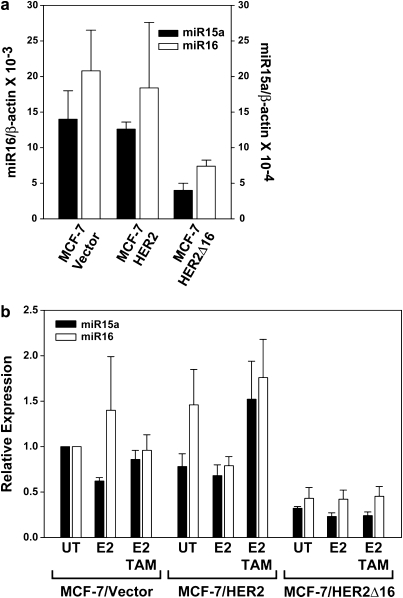
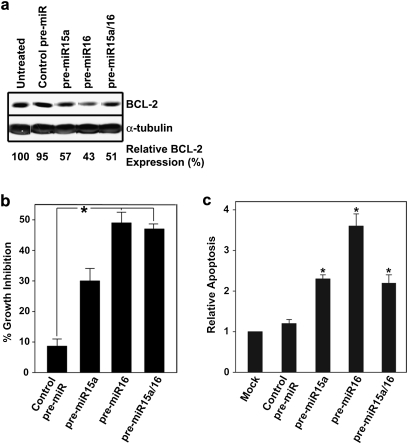
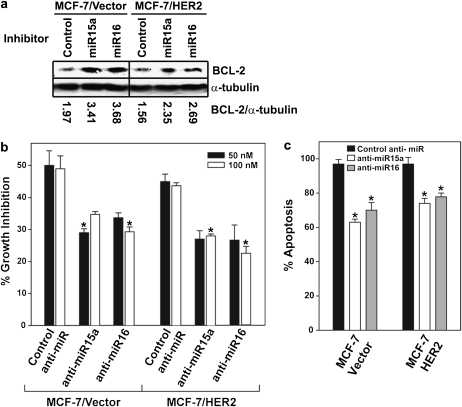
Tamoxifen is the most commonly prescribed therapy for patients with estrogen receptor (ER)α-positive breast tumors. Tumor resistance to tamoxifen remains a serious clinical problem especially in patients with tumors that also overexpress human epidermal growth factor receptor 2 (HER2). Current preclinical models of HER2 overexpression fail to recapitulate the clinical spectrum of endocrine resistance associated with HER2/ER-positive tumors. Here, we show that ectopic expression of a clinically important oncogenic isoform of HER2, HER2Δ16, which is expressed in >30% of ER-positive breast tumors, promotes tamoxifen resistance and estrogen independence of MCF-7 xenografts. MCF-7/HER2Δ16 cells evade tamoxifen through upregulation of BCL-2, whereas mediated suppression of BCL-2 expression or treatment of MCF-7/HER2Δ16 cells with the BCL-2 family pharmacological inhibitor ABT-737 restores tamoxifen sensitivity. Tamoxifen-resistant MCF-7/HER2Δ16 cells upregulate BCL-2 protein levels in response to suppressed ERα signaling mediated by estrogen withdrawal, tamoxifen treatment or fulvestrant treatment. In addition, HER2Δ16 expression results in suppression of BCL-2-targeting microRNAs miR-15a and miR-16. Reintroduction of miR-15a/16 reduced tamoxifen-induced BCL-2 expression and sensitized MCF-7/HER2Δ16 to tamoxifen. Conversely, inhibition of miR-15a/16 in tamoxifen-sensitive cells activated BCL-2 expression and promoted tamoxifen resistance. Our results suggest that HER2Δ16 expression promotes endocrine-resistant HER2/ERα-positive breast tumors and in contrast to wild-type HER2, preclinical models of HER2Δ16 overexpression recapitulate multiple phenotypes of endocrine-resistant human breast tumors. The mechanism of HER2Δ16 therapeutic evasion, involving tamoxifen-induced upregulation of BCL-2 and suppression of miR-15a/16, provides a template for unique therapeutic interventions combining tamoxifen with modulation of microRNAs and/or ABT-737-mediated BCL-2 inhibition and apoptosis.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
