

Elusive copy number variation in the mouse genome
- PMID: 20877625
- PMCID: PMC2943477
- DOI: 10.1371/journal.pone.0012839
Elusive copy number variation in the mouse genome
Abstract
Background: Array comparative genomic hybridization (aCGH) to detect copy number variants (CNVs) in mammalian genomes has led to a growing awareness of the potential importance of this category of sequence variation as a cause of phenotypic variation. Yet there are large discrepancies between studies, so that the extent of the genome affected by CNVs is unknown. We combined molecular and aCGH analyses of CNVs in inbred mouse strains to investigate this question.
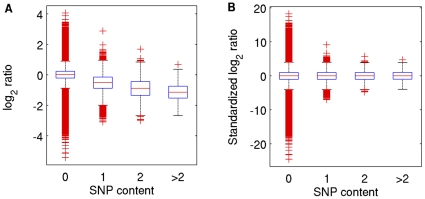
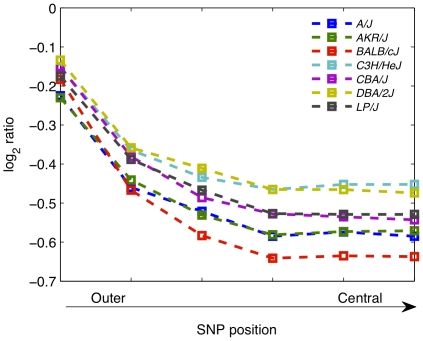
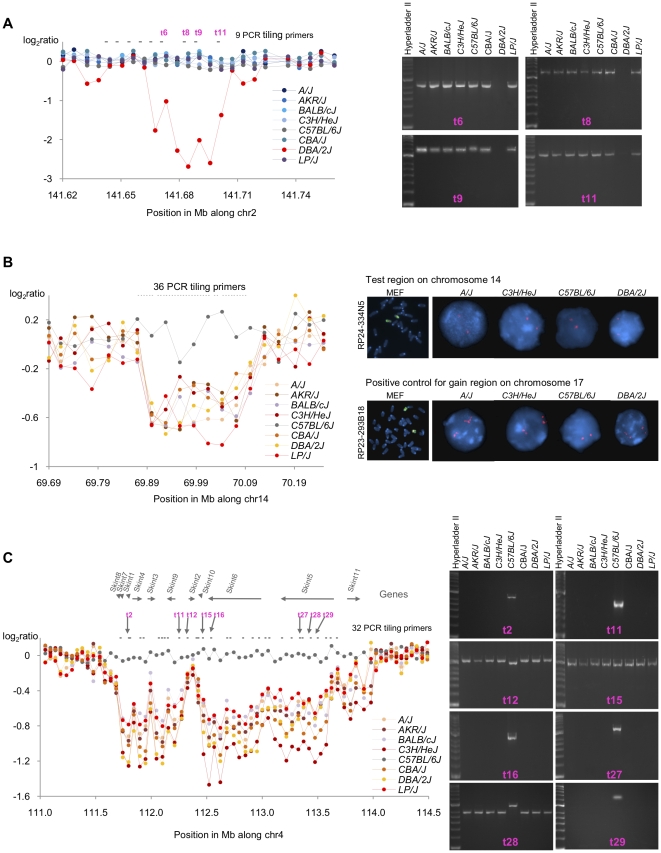
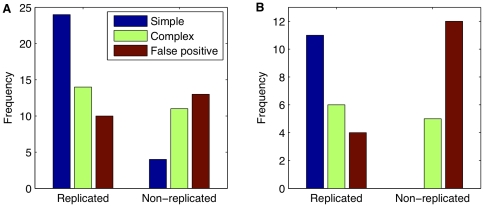
Principal findings: Using a 2.1 million probe array we identified 1,477 deletions and 499 gains in 7 inbred mouse strains. Molecular characterization indicated that approximately one third of the CNVs detected by the array were false positives and we estimate the false negative rate to be more than 50%. We show that low concordance between studies is largely due to the molecular nature of CNVs, many of which consist of a series of smaller deletions and gains interspersed by regions where the DNA copy number is normal.
Conclusions: Our results indicate that CNVs detected by arrays may be the coincidental co-localization of smaller CNVs, whose presence is more likely to perturb an aCGH hybridization profile than the effect of an isolated, small, copy number alteration. Our findings help explain the hitherto unexplored discrepancies between array-based studies of copy number variation in the mouse genome.
Conflict of interest statement
Figures
References
-
- Henrichsen CN, Vinckenbosch N, Zollner S, Chaignat E, Pradervand S, et al. Segmental copy number variation shapes tissue transcriptomes. Nat Genet. 2009;41:424–429. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
