

Type 2 Gaucher disease: phenotypic variation and genotypic heterogeneity
- PMID: 20880730
- PMCID: PMC3018671
- DOI: 10.1016/j.bcmd.2010.08.012
Type 2 Gaucher disease: phenotypic variation and genotypic heterogeneity
Abstract
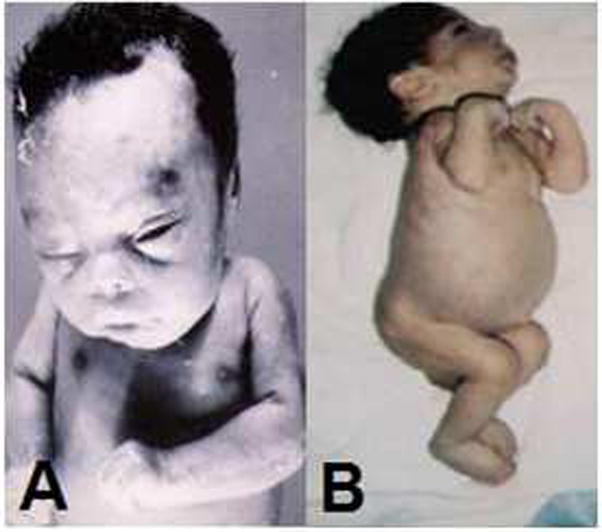
Gaucher disease (GD), the most common lysosomal storage disease, results from a deficiency of the lysosomal enzyme glucocerebrosidase. GD has been classified into 3 types, of which type 2 (the acute neuronopathic form) is the most severe, presenting pre- or perinatally, or in the first few months of life. Traditionally, type 2 GD was considered to have the most uniform clinical phenotype when compared to other GD subtypes. However, case studies over time have demonstrated that type 2 GD, like types 1 and 3, manifests with a spectrum of phenotypes. This review includes case reports that illustrate the broad range of clinical presentations encountered in type 2 GD, as well as a discussion of associated manifestations, pathological findings, diagnostic techniques, and a review of current therapies. While type 2 GD is generally associated with severe mutations in the glucocerebrosidase gene, there is also significant genotypic heterogeneity.
Published by Elsevier Inc.
Figures

References
-
- Knudson AG, Kaplan WD. Jewish Chronic Disease Hospital (Brooklyn New York N.Y.). Isaac Albert Research Institute. Genetics of the spingolipidoses. In: Aronson SM, Volk BW, editors. Cerebral sphingolipidoses; a symposium on Tay-Sachs’ disease and allied disorders. Academic Press; New York: 1962. p. xvii.p. 456.
-
- Fredrickson DS, Sloan HR. Glycosylceremide lipidoses: Gaucher’s disease. In: Stanbury JB, Wyngaarden JB, Fredrickson DS, editors. The metabolic basis of inherited disease. McGraw-Hill; New York: 1972. p. xiv.p. 1778.
-
- Oberling C, Woringer P. La maladie de Gaucher chez le nourrisson. Rev franc de pédiat. 1927;3:475–532.
-
- Beutler E, Grabowski GA. Gaucher disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 3635–3668.
-
- Tayebi N, Stone DL, Sidransky E. Type 2 gaucher disease: an expanding phenotype. Mol Genet Metab. 1999;68:209–19. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
