

Estrogens and development of pulmonary hypertension: interaction of estradiol metabolism and pulmonary vascular disease
- PMID: 20881610
- PMCID: PMC3027839
- DOI: 10.1097/FJC.0b013e3181f9ea8d
Estrogens and development of pulmonary hypertension: interaction of estradiol metabolism and pulmonary vascular disease
Abstract
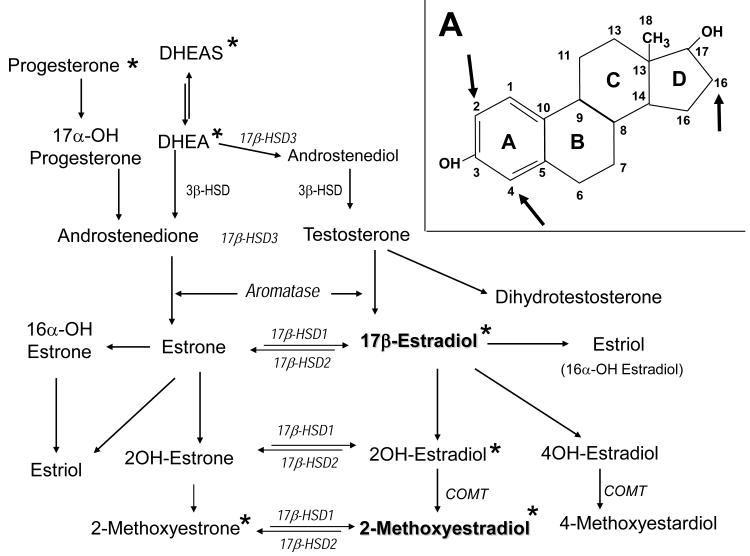
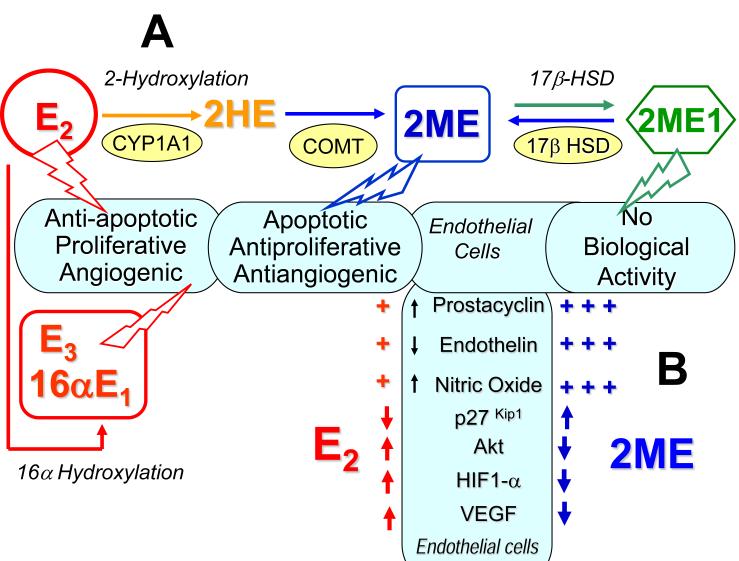
Severe pulmonary arterial hypertension (PAH) is characterized by clustered proliferation of endothelial cells (ECs) in the lumina of small size pulmonary arteries resulting in concentric obliteration of the lumina and formation of complex vascular structures known as plexiform lesions. This debilitating disease occurs more frequently in women, yet both animal studies in classical models of PAH and limited clinical data suggest protective effects of estrogens: the estrogen paradox in pulmonary hypertension. Little is known about the role of estrogens in PAH, but one line of evidence strongly suggests that the vascular protective effects of 17β-estradiol (estradiol; E2) are mediated largely by its downstream metabolites. Estradiol is metabolized to 2-hydroxyestradiol (2HE) by CYP1A1/CYP1B1, and 2HE is converted to 2-methoxyestradiol (2ME) by catechol-O-methyl transferase. 2ME is extensively metabolized to 2-methoxyestrone, a metabolite that lacks biologic activity, but which may be converted back to 2ME. 2ME has no estrogenic activity, and its effects are mediated by estrogen receptors–independent mechanism(s). Notably, in systemic and pulmonary vascular ECs, smooth muscle cells, and fibroblasts, 2ME exerts stronger antimitotic effects than E2 itself. E2 and 2ME, despite having similar effects on other cardiovascular cells, have opposing effects on ECs; that is, in ECs, E2 is promitogenic, proangiogenic, and antiapoptotic, whereas 2ME is antimitogenic, antiangiogenic, and proapoptotic. This may have significant ramifications in severe PAH that involves uncontrolled proliferation of monoclonal apoptosis-resistant ECs. Based on its cellular effects, 2ME should be expected to attenuate the progression of disease and provide protection in severe PAH. In contrast, E2, due to its mitogenic, angiogenic, and antiapoptotic effects (otherwise desirable in normal quiescent ECs), may even adversely affect endothelial remodeling in PAH, and this may be even more significant if the E2's effects on injured endothelium are not opposed by 2ME (eg, in the event of reduced E2 conversion to 2ME due to hypoxia, inflammation, drugs, environmental factors, or genetic polymorphism of metabolizing enzymes). This review focuses on the effects of estrogens and their metabolites on pulmonary vascular pathobiology and the development of experimental PAH and offers potential explanation for the estrogen paradox in PAH. Furthermore, we propose that unbalanced estradiol metabolism may lead to the development of PAH. Recent animal data and studies in patients with PAH support this concept.
Figures
References
-
- Badesh BD, Champion HC, Gomez-Sanchez MA, Hoper M, Loyd J, Manes A, McGoon MD, Naeije R, Olshewski H, Oudiz R, Torbicki A. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S55–S56. - PubMed
-
- Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, Elliott CG, Gaine S, Gladwin MT, Jing ZC, Krowka MJ, Langleben D, Nakanishi N, Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43–S54. - PubMed
-
- Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, Dupuis J, Long CS, Rubin LJ, Smart FW, Suzuki YJ, Gladwin M, Denholm EM, Gail DB. Right ventricular function and failure: report of a National Heart, Lung and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation. 2006;114:1883–1891. - PubMed
-
- Thenappan T, Shah SJ, Rich S, Gomberg-Maitland M. A USA-based registry for pulmonary arterial hypertension: 1982-2006. Eur Res J. 2007;30:1103–1110. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
