

Catalytic function of PLA2G6 is impaired by mutations associated with infantile neuroaxonal dystrophy but not dystonia-parkinsonism
- PMID: 20886109
- PMCID: PMC2944820
- DOI: 10.1371/journal.pone.0012897
Catalytic function of PLA2G6 is impaired by mutations associated with infantile neuroaxonal dystrophy but not dystonia-parkinsonism
Abstract
Background: Mutations in the PLA2G6 gene have been identified in autosomal recessive neurodegenerative diseases classified as infantile neuroaxonal dystrophy (INAD), neurodegeneration with brain iron accumulation (NBIA), and dystonia-parkinsonism. These clinical syndromes display two significantly different disease phenotypes. NBIA and INAD are very similar, involving widespread neurodegeneration that begins within the first 1-2 years of life. In contrast, patients with dystonia-parkinsonism present with a parkinsonian movement disorder beginning at 15 to 30 years of age. The PLA2G6 gene encodes the PLA2G6 enzyme, also known as group VIA calcium-independent phospholipase A(2), which has previously been shown to hydrolyze the sn-2 acyl chain of phospholipids, generating free fatty acids and lysophospholipids.
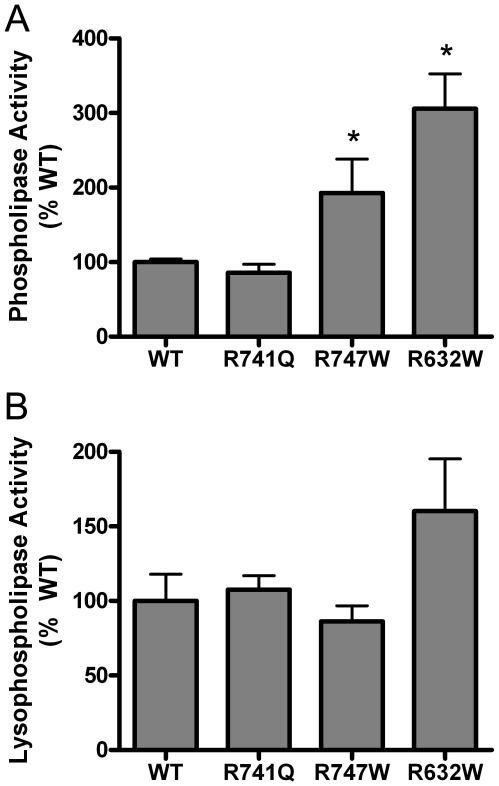
Methodology/principal findings: We produced purified recombinant wildtype (WT) and mutant human PLA2G6 proteins and examined their catalytic function using in vitro assays with radiolabeled lipid substrates. We find that human PLA2G6 enzyme hydrolyzes both phospholipids and lysophospholipids, releasing free fatty acids. Mutations associated with different disease phenotypes have different effects on catalytic activity. Mutations associated with INAD/NBIA cause loss of enzyme activity, with mutant proteins exhibiting less than 20% of the specific activity of WT protein in both lysophospholipase and phospholipase assays. In contrast, mutations associated with dystonia-parkinsonism do not impair catalytic activity, and two mutations produce a significant increase in specific activity for phospholipid but not lysophospholipid substrates.
Conclusions/significance: These results indicate that different alterations in PLA2G6 function produce the different disease phenotypes of NBIA/INAD and dystonia-parkinsonism. INAD/NBIA is caused by loss of the ability of PLA2G6 to catalyze fatty acid release from phospholipids, which predicts accumulation of PLA2G6 phospholipid substrates and provides a mechanistic explanation for the accumulation of membranes in neuroaxonal spheroids previously observed in histopathological studies of INAD/NBIA. In contrast, dystonia-parkinsonism mutations do not appear to directly impair catalytic function, but may modify substrate preferences or regulatory mechanisms for PLA2G6.
Conflict of interest statement
Figures
References
-
- Sina F, Shojaee S, Elahi E, Paisan-Ruiz C. R632W mutation in PLA2G6 segregates with dystonia-parkinsonism in a consanguineous Iranian family. Eur J Neurol. 2009;16:101–104. - PubMed
-
- Kurian MA, Morgan NV, MacPherson L, Foster K, Peake D, et al. Phenotypic spectrum of neurodegeneration associated with mutations in the PLA2G6 gene (PLAN). Neurology. 2008;70:1623–1629. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
