

Protein homeostasis in models of aging and age-related conformational disease
- PMID: 20886762
- PMCID: PMC3402352
- DOI: 10.1007/978-1-4419-7002-2_11
Protein homeostasis in models of aging and age-related conformational disease
Abstract
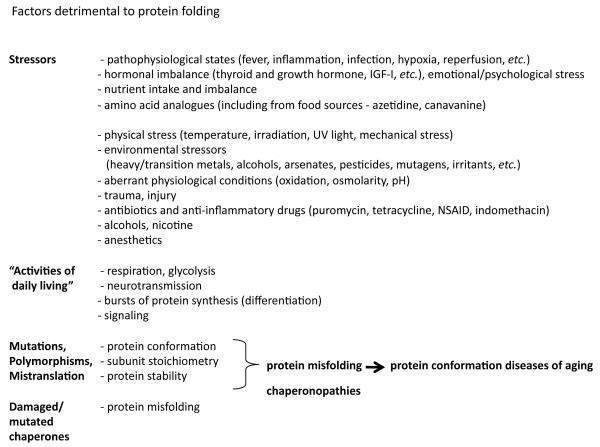
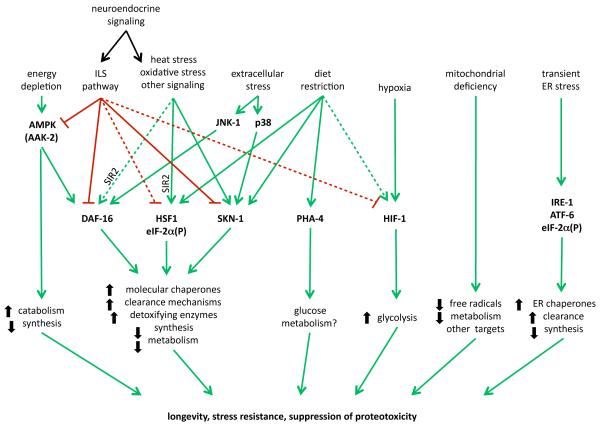
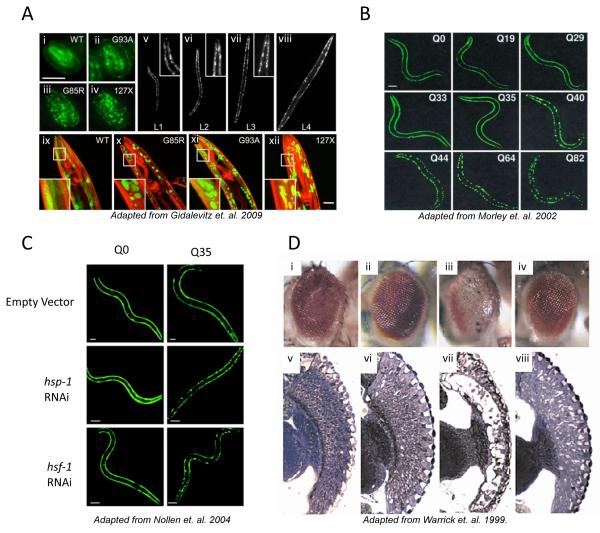
The stability of the proteome is crucial to the health of the cell, and contributes significantly to the lifespan of the organism. Aging and many age-related diseases have in common the expression of misfolded and damaged proteins. The chronic expression of damaged proteins during disease can have devastating consequences on protein homeostasis (proteostasis), resulting in disruption ofnumerous biological processes. This chapter discusses our current understanding of the various contributors to protein misfolding, and the mechanisms by which misfolding, and accompanied aggregation/toxicity, is accelerated by stress and aging. Invertebrate models have been instrumental in studying the processes related to aggregation and toxicity of disease-associated proteins and how dysregulation ofproteostasis leads to neurodegenerative diseases of aging.
Figures
References
-
- Rutherford SL, Lindquist S. Hsp90 as a capacitor for morphological evolution. Nature. 1998;396:336–342. - PubMed
-
- Stevens FJ, Argon Y. Pathogenic light chains and the B-cell repertoire. Immunol Today. 1999;20:451–457. - PubMed
-
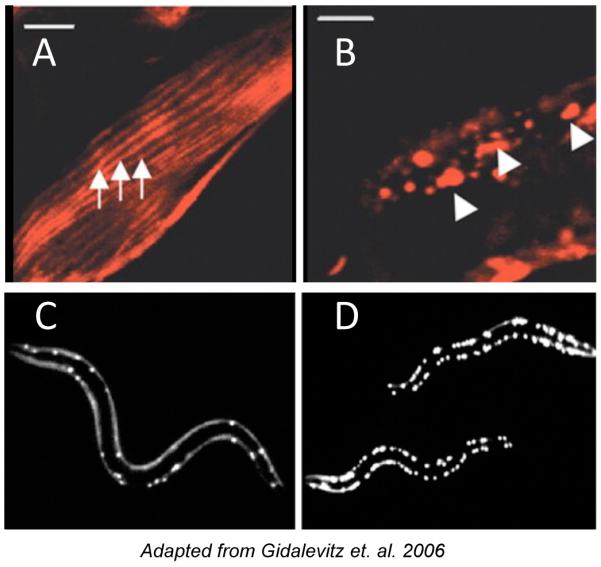
- Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–1474. - PubMed
-
- Michels AA, et al. Hsp70 and Hsp40 chaperone activities in the cytoplasm and the nucleus of mammalian cells. J Biol Chem. 1997;272:33283–33289. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
