

The pathogenesis of sepsis
- PMID: 20887193
- PMCID: PMC3684427
- DOI: 10.1146/annurev-pathol-011110-130327
The pathogenesis of sepsis
Abstract
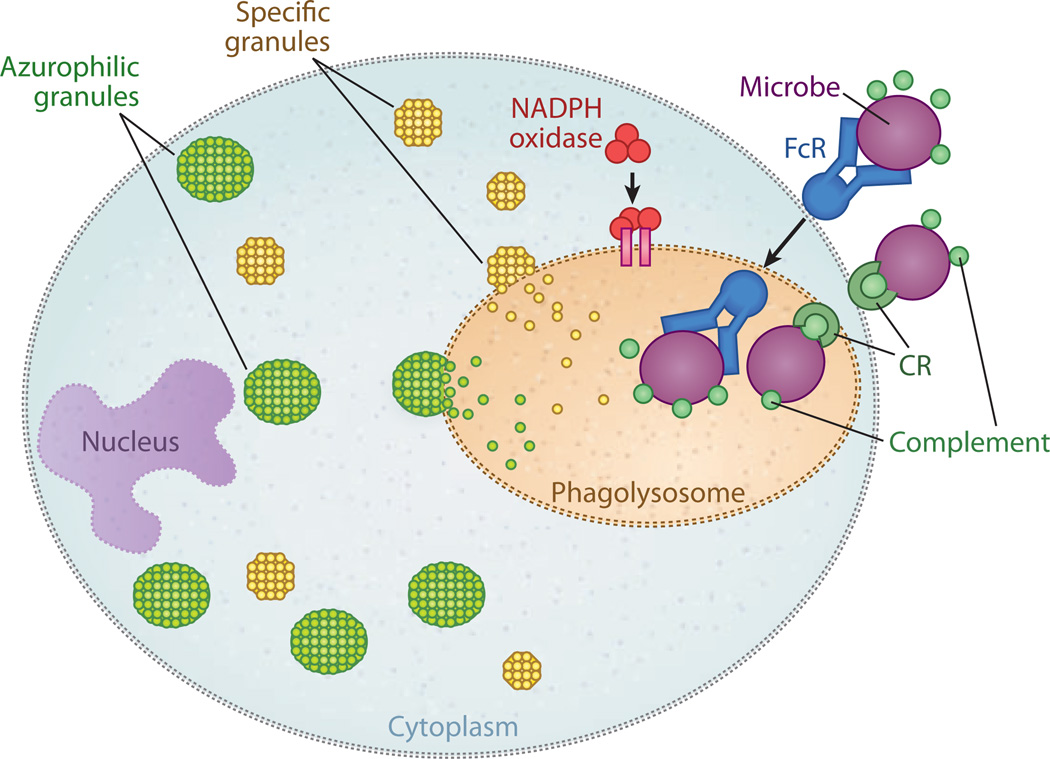
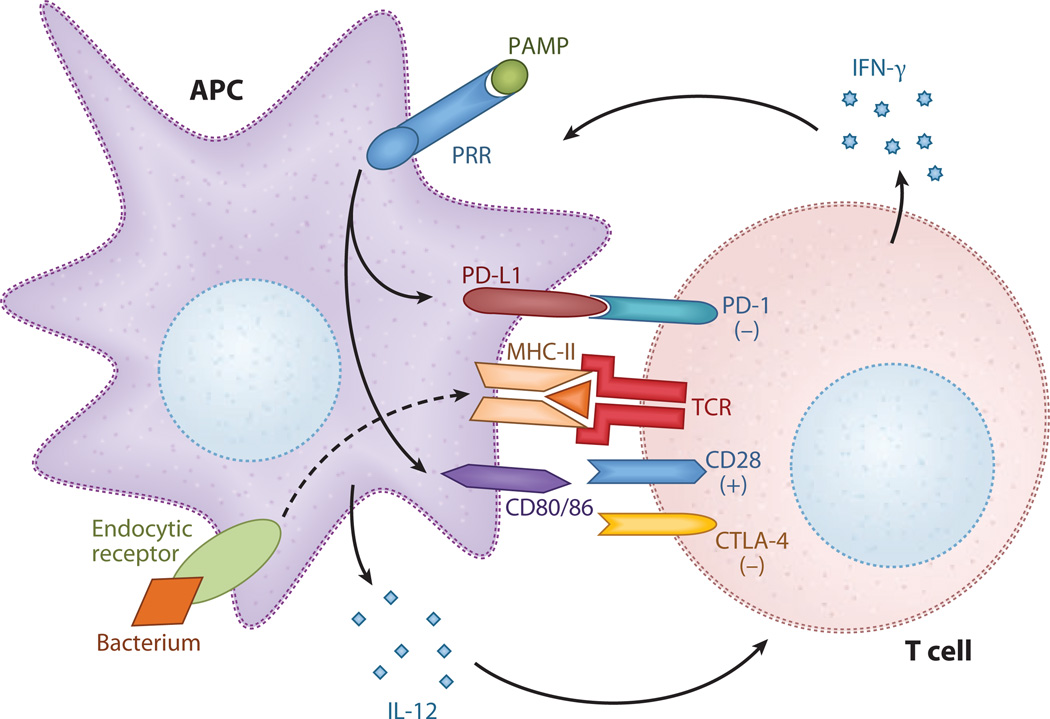
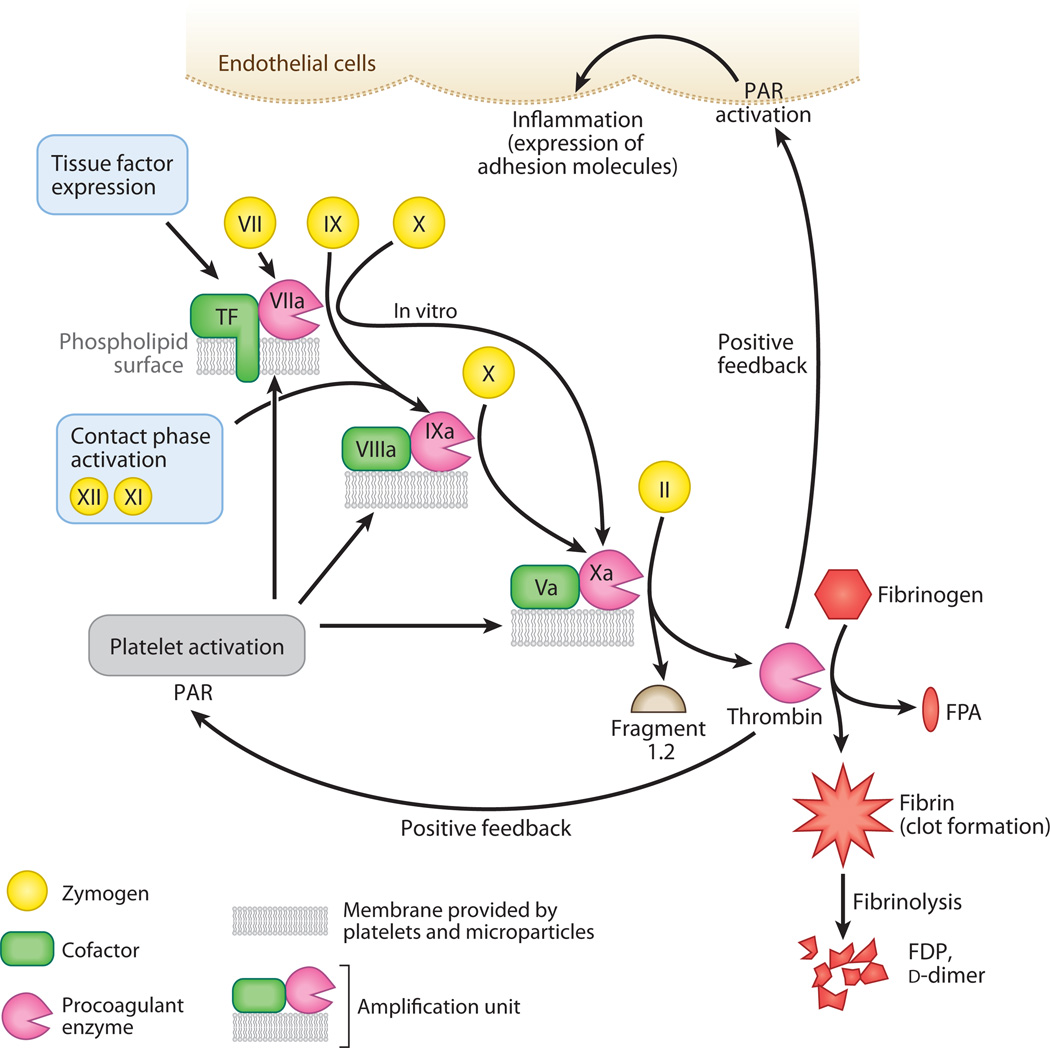
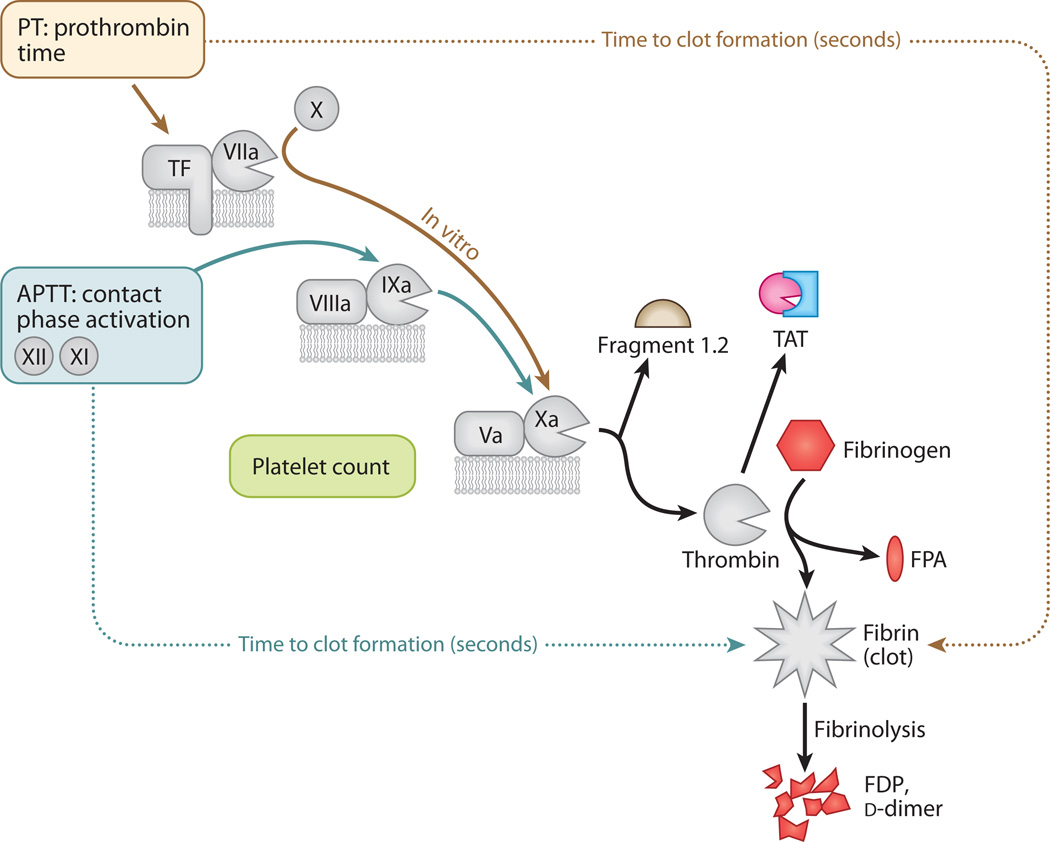
Sepsis is a serious clinical condition that represents a patient's response to a severe infection and has a very high mortality rate. Normal immune and physiologic responses eradicate pathogens, and the pathophysiology of sepsis is due to the inappropriate regulation of these normal reactions. In an ideal scenario, the first pathogen contact with the inflammatory system should eliminate the microbe and quickly return the host to homeostasis. The septic response may accelerate due to continued activation of neutrophils and macrophages/monocytes. Upregulation of lymphocyte costimulatory molecules and rapid lymphocyte apoptosis, delayed apoptosis of neutrophils, and enhanced necrosis of cells/tissues also contribute to the pathogenesis of sepsis. The coagulation system is closely tied to the inflammatory response, with cross talk between the two systems driving the dysregulated response. Biomarkers may be used to help diagnose patients with sepsis, and they may also help to identify patients who would benefit from immunomodulatory therapies.
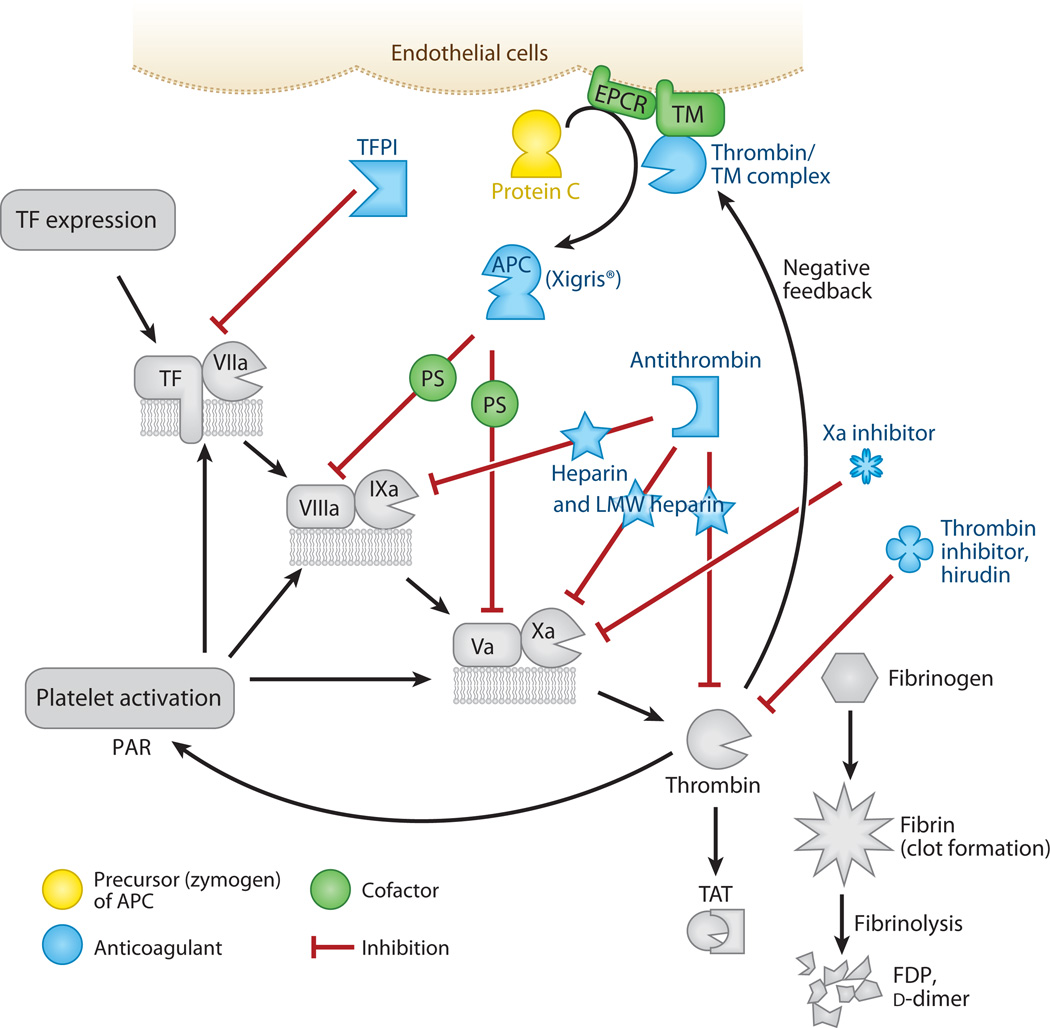
Figures
References
-
- Benoit M, Desnues B, Mege JL. Macrophage polarization in bacterial infections. J. Immunol. 2008;181:3733–3739. - PubMed
-
- Urban CF, Lourido S, Zychlinsky A. How do microbes evade neutrophil killing? Cell Microbiol. 2006;8:1687–1696. - PubMed
-
- Le Tulzo Y, Pangault C, Gacouin A, Guilloux V, Tribut O, et al. Early circulating lymphocyte apoptosis in human septic shock is associated with poor outcome. Shock. 2002;18:487–494. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
