

Mutations in SCARF2 are responsible for Van Den Ende-Gupta syndrome
- PMID: 20887961
- PMCID: PMC2948800
- DOI: 10.1016/j.ajhg.2010.09.005
Mutations in SCARF2 are responsible for Van Den Ende-Gupta syndrome
Abstract
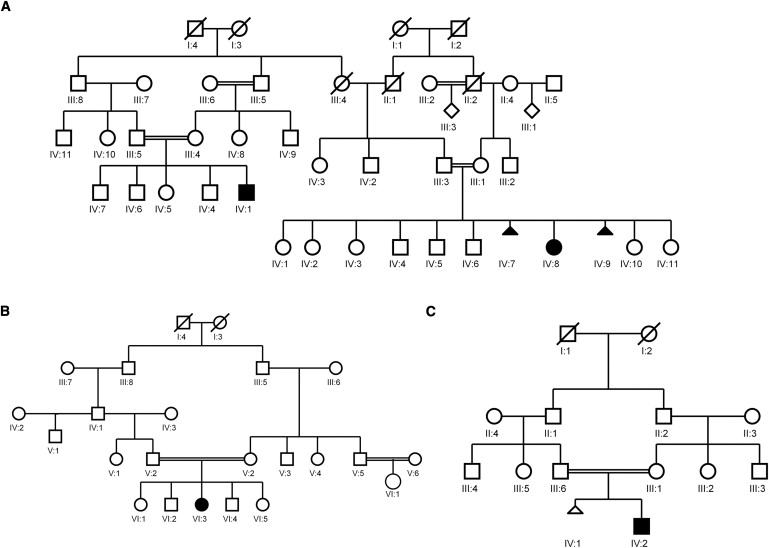
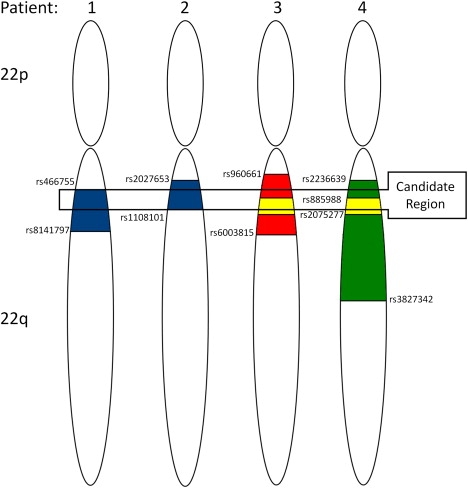
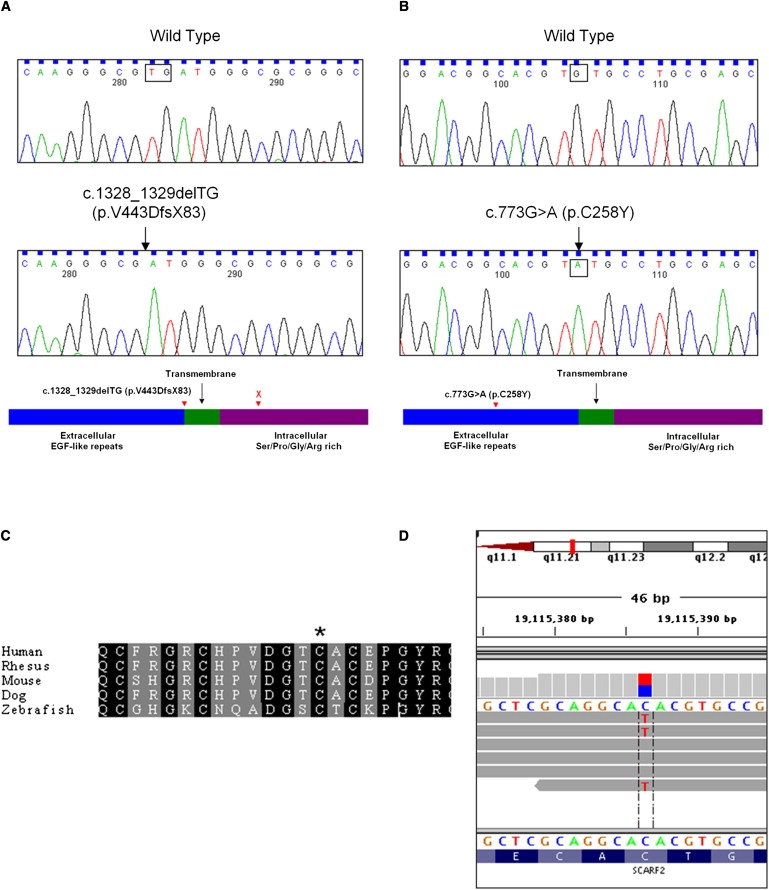
Van Den Ende-Gupta syndrome (VDEGS) is an extremely rare autosomal-recessive disorder characterized by distinctive craniofacial features, which include blepharophimosis, malar and/or maxillary hypoplasia, a narrow and beaked nose, and an everted lower lip. Other features are arachnodactyly, camptodactyly, peculiar skeletal abnormalities, and normal development and intelligence. We present molecular data on four VDEGS patients from three consanguineous Qatari families belonging to the same highly inbred Bedouin tribe. The patients were genotyped with SNP microarrays, and a 2.4 Mb homozygous region was found on chromosome 22q11 in an area overlapping the DiGeorge critical region. This region contained 44 genes, including SCARF2, a gene that is expressed during development in a number of mouse tissues relevant to the symptoms described above. Sanger sequencing identified a missense change, c.773G>A (p.C258Y), in exon 4 in the two closely related patients and a 2 bp deletion in exon 8, c.1328_1329delTG (p.V443DfsX83), in two unrelated individuals. In parallel with the candidate gene approach, complete exome sequencing was used to confirm that SCARF2 was the gene responsible for VDEGS. SCARF2 contains putative epidermal growth factor-like domains in its extracellular domain, along with a number of positively charged residues in its intracellular domain, indicating that it may be involved in intracellular signaling. However, the function of SCARF2 has not been characterized, and this study reports that phenotypic effects can be associated with defects in the scavenger receptor F family of genes.
Copyright © 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Sclerocornea in a patient with van den Ende-Gupta syndrome homozygous for a SCARF2 microdeletion.Am J Med Genet A. 2014 May;164A(5):1170-4. doi: 10.1002/ajmg.a.36425. Epub 2014 Jan 29. Am J Med Genet A. 2014. PMID: 24478002 Free PMC article.
-
Inclusion of joint laxity, recurrent patellar dislocation, and short distal ulnae as a feature of Van Den Ende-Gupta syndrome: a case report.BMC Med Genet. 2018 Jan 30;19(1):18. doi: 10.1186/s12881-018-0531-y. BMC Med Genet. 2018. PMID: 29378527 Free PMC article.
-
Further delineation of van den Ende-Gupta syndrome: Genetic heterogeneity and overlap with congenital heart defects and skeletal malformations syndrome.Am J Med Genet A. 2021 Jul;185(7):2136-2149. doi: 10.1002/ajmg.a.62194. Epub 2021 Mar 30. Am J Med Genet A. 2021. PMID: 33783941
-
Exploring scavenger receptor class F member 2 and the importance of scavenger receptor family in prediagnostic diseases.Toxicol Res. 2023 Apr 14;39(3):341-353. doi: 10.1007/s43188-023-00176-2. eCollection 2023 Jul. Toxicol Res. 2023. PMID: 37398563 Free PMC article. Review.
-
Identification of a novel variant of SCARF2 in a Jordanian family with a van den Ende-Gupta Syndrome and literature review.Clin Dysmorphol. 2022 Jul 1;31(3):157-161. doi: 10.1097/MCD.0000000000000415. Epub 2022 Mar 7. Clin Dysmorphol. 2022. PMID: 35256560 Review. No abstract available.
Cited by
-
Unmasking of a Recessive SCARF2 Mutation by a 22q11.12 de novo Deletion in a Patient with Van den Ende-Gupta Syndrome.Mol Syndromol. 2010;1(5):239-245. doi: 10.1159/000328135. Epub 2011 May 18. Mol Syndromol. 2010. PMID: 22140376 Free PMC article.
-
Sclerocornea in a patient with van den Ende-Gupta syndrome homozygous for a SCARF2 microdeletion.Am J Med Genet A. 2014 May;164A(5):1170-4. doi: 10.1002/ajmg.a.36425. Epub 2014 Jan 29. Am J Med Genet A. 2014. PMID: 24478002 Free PMC article.
-
Incidence of the 22q11.2 deletion in a large cohort of miscarriage samples.Mol Cytogenet. 2017 Mar 9;10:6. doi: 10.1186/s13039-017-0308-6. eCollection 2017. Mol Cytogenet. 2017. PMID: 28293297 Free PMC article.
-
Scavenger receptor class F member 2 (SCARF2) as a novel therapeutic target in glioblastoma.Toxicol Res. 2022 Feb 25;38(2):249-256. doi: 10.1007/s43188-022-00125-5. eCollection 2022 Apr. Toxicol Res. 2022. PMID: 35419275 Free PMC article.
-
Inclusion of joint laxity, recurrent patellar dislocation, and short distal ulnae as a feature of Van Den Ende-Gupta syndrome: a case report.BMC Med Genet. 2018 Jan 30;19(1):18. doi: 10.1186/s12881-018-0531-y. BMC Med Genet. 2018. PMID: 29378527 Free PMC article.
References
-
- van den Ende J.J., van Bever Y., Rodini E.S., Richieri-Costa A. Marden-Walker-like syndrome without psychomotor retardation: Report of a Brazilian girl born to consanguineous parents. Am. J. Med. Genet. 1992;42:467–469. - PubMed
-
- Bistritzer T., Fried K., Lahat E., Dvir M., Goldberg M. Congenital contractural arachnodactyly in two double second cousins: Possible homozygosity. Clin. Genet. 1993;44:15–19. - PubMed
-
- Carr C.W., Carron J.D., Lachman R.S., Abdul-Rahman O.A. Van den Ende-Gupta syndrome: Laryngeal abnormalities in two siblings. Am. J. Med. Genet. A. 2007;143A:2706–2711. - PubMed
-
- Leal G.F., Silva E.O. van den Ende-Gupta syndrome: Evidence for genetic heterogeneity. Am. J. Med. Genet. A. 2009;149A:1293–1295. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
