

Blocking the mitochondrial apoptotic pathway preserves motor neuron viability and function in a mouse model of amyotrophic lateral sclerosis
- PMID: 20890041
- PMCID: PMC2947232
- DOI: 10.1172/JCI42986
Blocking the mitochondrial apoptotic pathway preserves motor neuron viability and function in a mouse model of amyotrophic lateral sclerosis
Abstract
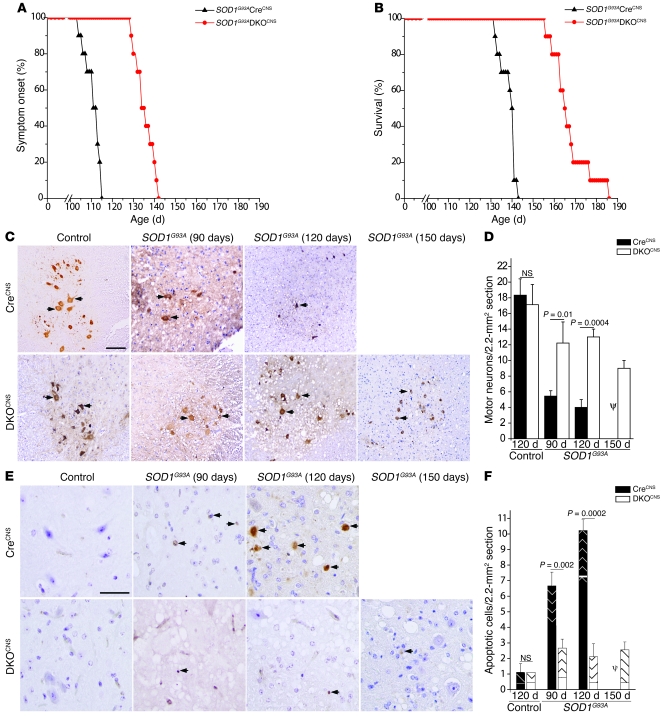
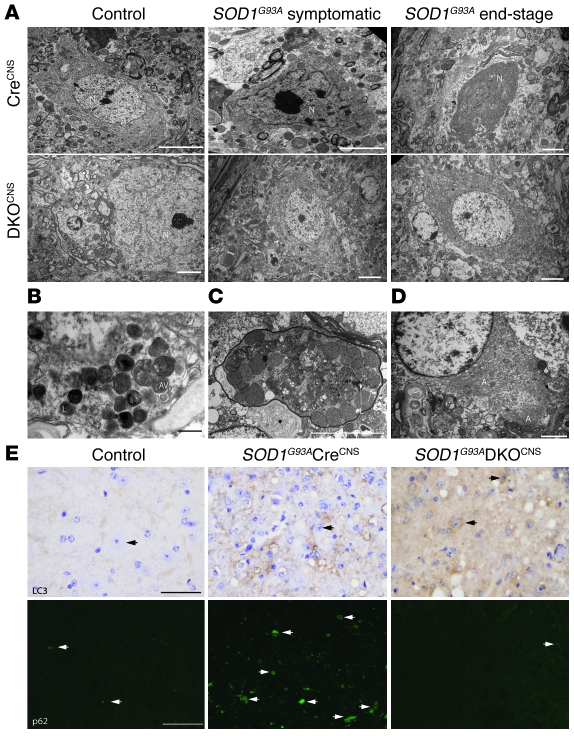
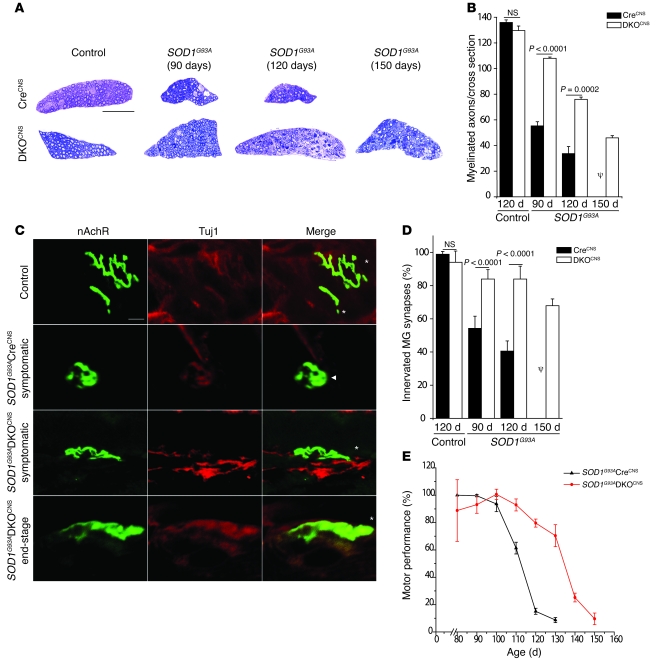
Apoptosis of motor neurons is a well-documented feature in amyotrophic lateral sclerosis (ALS) and related motor neuron diseases (MNDs). However, the role of apoptosis in the pathogenesis of these diseases remains unresolved. One possibility is that the affected motor neurons only succumb to apoptosis once they have exhausted functional capacity. If true, blocking apoptosis should confer no therapeutic benefit. To directly investigate this idea, we tested whether tissue-specific deletion in the mouse CNS of BCL2-associated X protein (BAX) and BCL2-homologous antagonist/killer (BAK), 2 proapoptotic BCL-2 family proteins that together represent an essential gateway to the mitochondrial apoptotic pathway, would protect against motor neuron degeneration. We found that neuronal deletion of Bax and Bak in a mouse model of familial ALS not only halted neuronal loss, but prevented axonal degeneration, symptom onset, weight loss, and paralysis and extended survival. These results show that motor neurons damaged in ALS activate the mitochondrial apoptotic pathway early in the disease process and that apoptotic signaling directly contributes to neuromuscular degeneration and neuronal dysfunction. Hence, inhibiting apoptosis upstream of mitochondrial permeabilization represents a possible therapeutic strategy for preserving functional motor neurons in ALS and other MNDs.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
