

Mutations in the SPTLC2 subunit of serine palmitoyltransferase cause hereditary sensory and autonomic neuropathy type I
- PMID: 20920666
- PMCID: PMC2948807
- DOI: 10.1016/j.ajhg.2010.09.010
Mutations in the SPTLC2 subunit of serine palmitoyltransferase cause hereditary sensory and autonomic neuropathy type I
Abstract
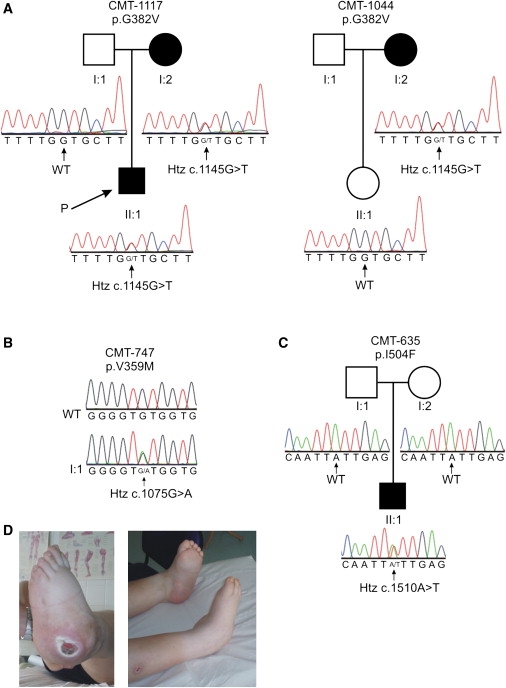
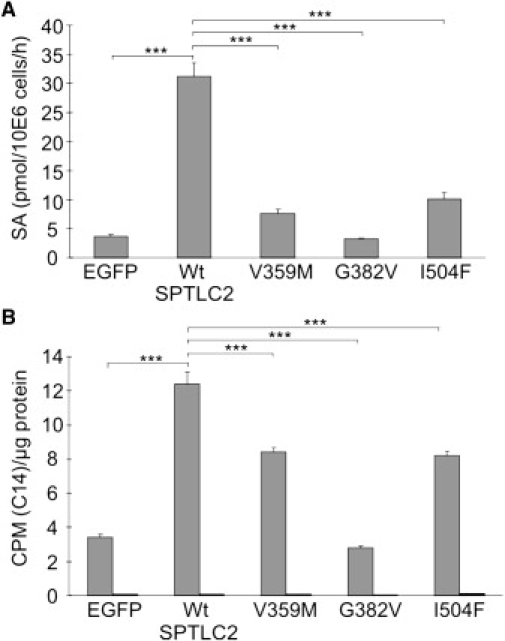
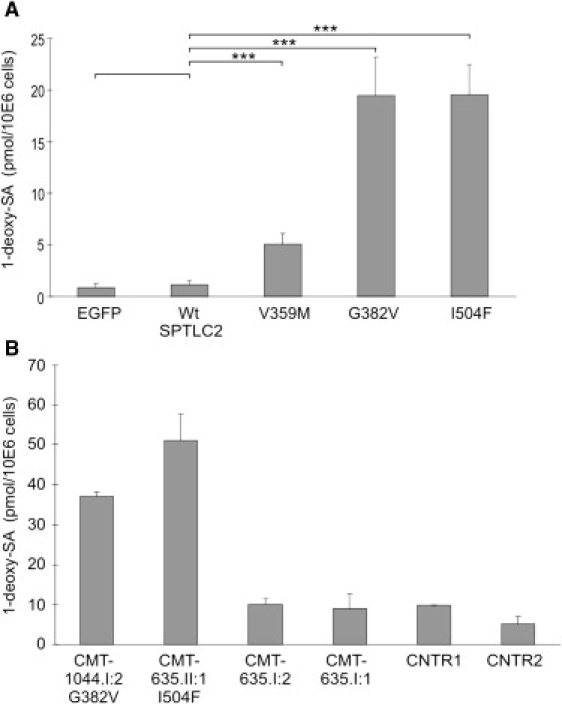
Hereditary sensory and autonomic neuropathy type I (HSAN-I) is an axonal peripheral neuropathy associated with progressive distal sensory loss and severe ulcerations. Mutations in the first subunit of the enzyme serine palmitoyltransferase (SPT) have been associated with HSAN-I. The SPT enzyme catalyzes the first and rate-limiting step in the de novo sphingolipid synthesis pathway. However, different studies suggest the implication of other genes in the pathology of HSAN-I. Therefore, we screened the two other known subunits of SPT, SPTLC2 and SPTLC3, in a cohort of 78 HSAN patients. No mutations were found in SPTLC3, but we identified three heterozygous missense mutations in the SPTLC2 subunit of SPT in four families presenting with a typical HSAN-I phenotype. We demonstrate that these mutations result in a partial to complete loss of SPT activity in vitro and in vivo. Moreover, they cause the accumulation of the atypical and neurotoxic sphingoid metabolite 1-deoxy-sphinganine. Our findings extend the genetic heterogeneity in HSAN-I and enlarge the group of HSAN neuropathies associated with SPT defects. We further show that HSAN-I is consistently associated with an increased formation of the neurotoxic 1-deoxysphinganine, suggesting a common pathomechanism for HSAN-I.
Copyright © 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures


References
-
- Dyck P.J., Chance P., Lebo R., Carney J.A. Hereditary motor and sensory neuropathies. In: Dyck P.J., Thomas P.K., Griffin J.W., Low P.A., Poduslo J.F., editors. Peripheral neuropathy. Third Edition. W.B. Saunders; Philadelphia: 1993. pp. 1065–1093.
-
- Auer-Grumbach M. Hereditary sensory neuropathies. Drugs Today (Barc) 2004;40:385–394. - PubMed
-
- Verpoorten N., De Jonghe P., Timmerman V. Disease mechanisms in hereditary sensory and autonomic neuropathies. Neurobiol. Dis. 2006;21:247–255. - PubMed
-
- Bejaoui K., Wu C., Scheffler M.D., Haan G., Ashby P., Wu L., de Jong P., Brown R.H., Jr. SPTLC1 is mutated in hereditary sensory neuropathy, type 1. Nat. Genet. 2001;27:261–262. - PubMed
-
- Dawkins J.L., Hulme D.J., Brahmbhatt S.B., Auer-Grumbach M., Nicholson G.A. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat. Genet. 2001;27:309–312. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
