

Characterization of Puma-dependent and Puma-independent neuronal cell death pathways following prolonged proteasomal inhibition
- PMID: 20921277
- PMCID: PMC2976426
- DOI: 10.1128/MCB.00575-10
Characterization of Puma-dependent and Puma-independent neuronal cell death pathways following prolonged proteasomal inhibition
Abstract
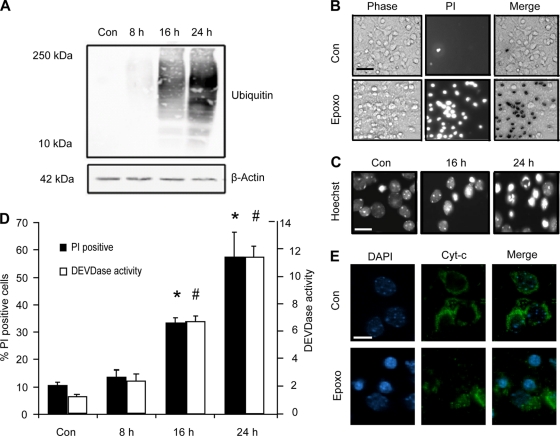
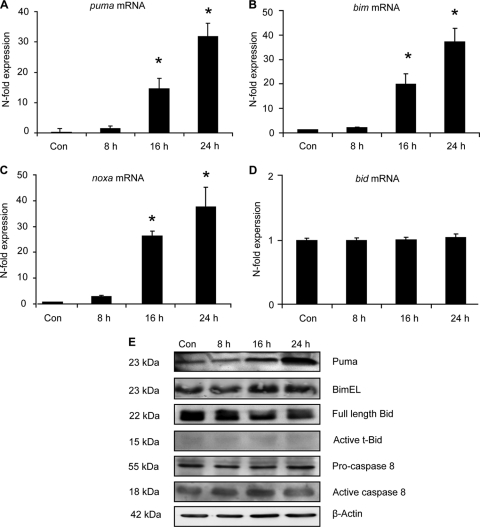
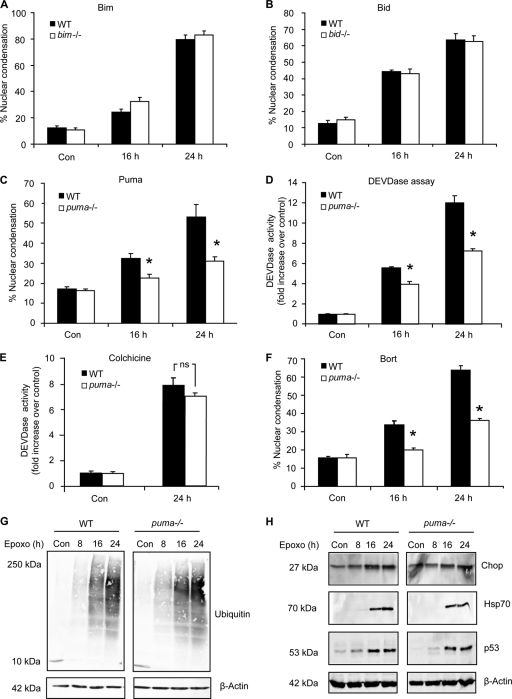
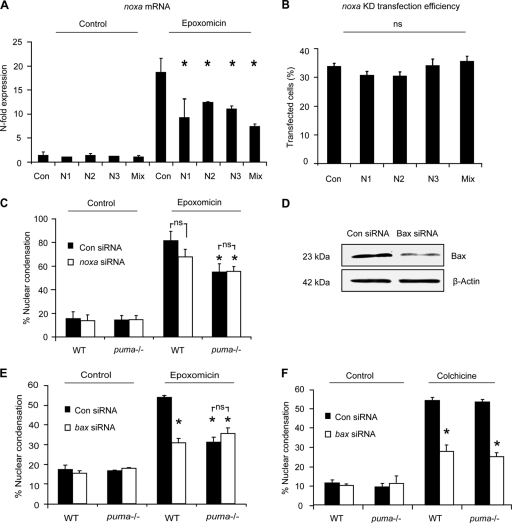
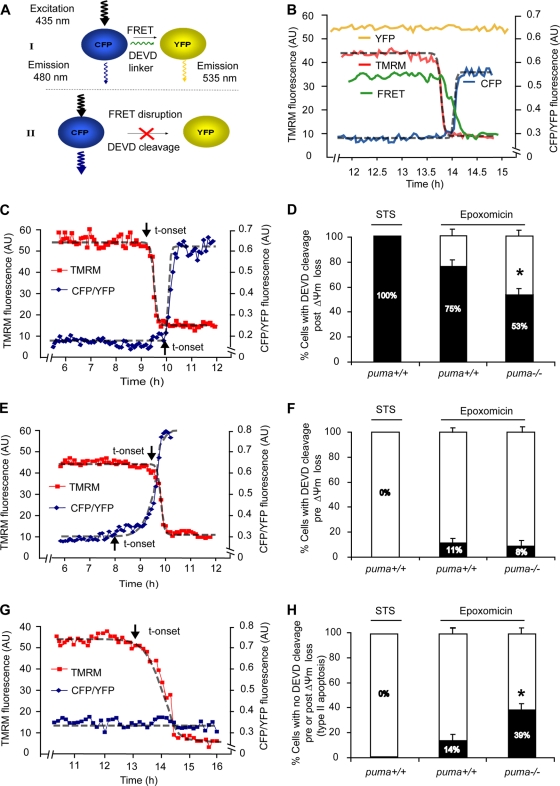
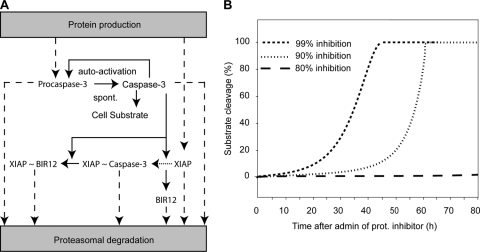
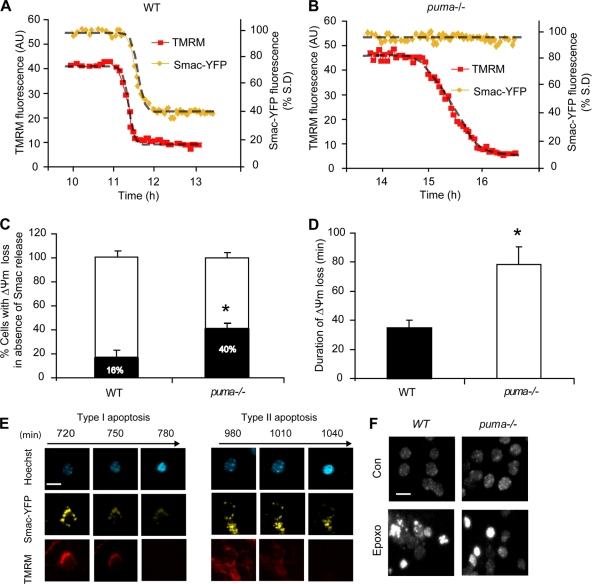
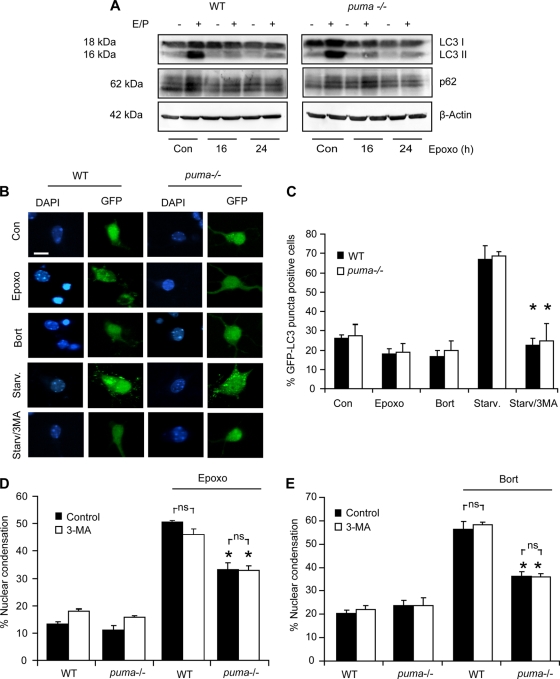
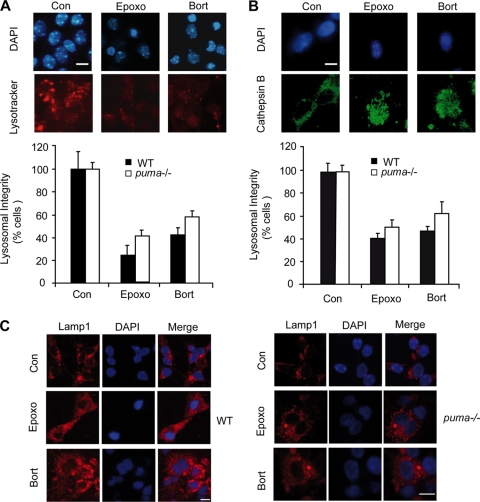
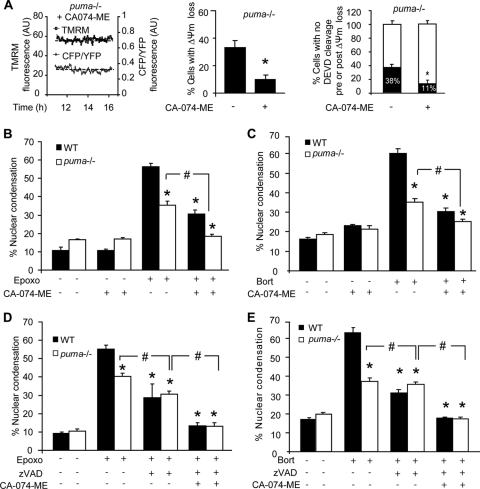
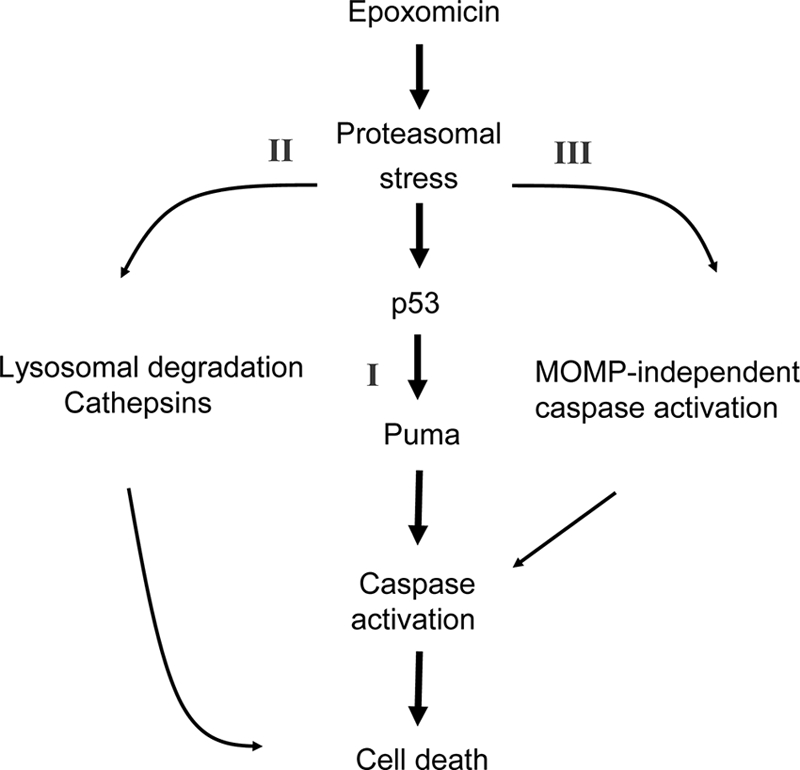
Proteasomal stress and the accumulation of polyubiquitinated proteins are key features of numerous neurodegenerative disorders. Previously we demonstrated that stabilization of p53 and activation of its target gene, puma (p53-upregulated mediator of apoptosis), mediated proteasome inhibitor-induced apoptosis in cancer cells. Here we demonstrated that Puma also contributed to proteasome inhibitor-induced apoptosis in mouse neocortical neurons. Although protection afforded by puma gene deletion was incomplete, we found little evidence indicating contributions from other proapoptotic BH3-only proteins. Attenuation of bax expression did not further reduce Puma-independent apoptosis, suggesting that pathways other than the mitochondrial apoptosis pathway were activated. Real-time imaging experiments in wild-type and puma-deficient neurons using a fluorescence resonance energy transfer (FRET)-based caspase sensor confirmed the involvement of a second cell death pathway characterized by caspase activation prior to mitochondrial permeabilization and, more prominently, a third, caspase-independent and Puma-independent pathway characterized by rapid cell shrinkage and nuclear condensation. This pathway involved lysosomal permeabilization in the absence of autophagy activation and was sensitive to cathepsin but not autophagy inhibition. Our data demonstrate that proteasomal stress activates distinct cell death pathways in neurons, leading to both caspase-dependent and caspase-independent apoptosis, and demonstrate independent roles for Puma and lysosomal permeabilization in this model.
Figures
Similar articles
-
Bcl-2 homology domain 3-only proteins Puma and Bim mediate the vulnerability of CA1 hippocampal neurons to proteasome inhibition in vivo.Eur J Neurosci. 2011 Feb;33(3):401-8. doi: 10.1111/j.1460-9568.2010.07538.x. Epub 2010 Dec 31. Eur J Neurosci. 2011. PMID: 21198986
-
The proapoptotic BH3-only, Bcl-2 family member, Puma is critical for acute ethanol-induced neuronal apoptosis.J Neuropathol Exp Neurol. 2009 Jul;68(7):747-56. doi: 10.1097/NEN.0b013e3181a9d524. J Neuropathol Exp Neurol. 2009. PMID: 19535997 Free PMC article.
-
BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program.Science. 2010 Dec 3;330(6009):1390-3. doi: 10.1126/science.1190217. Science. 2010. PMID: 21127253 Free PMC article.
-
PUMA, a potent killer with or without p53.Oncogene. 2008 Dec;27 Suppl 1(Suppl 1):S71-83. doi: 10.1038/onc.2009.45. Oncogene. 2008. PMID: 19641508 Free PMC article. Review.
-
PUMA, a critical mediator of cell death--one decade on from its discovery.Cell Mol Biol Lett. 2012 Dec;17(4):646-69. doi: 10.2478/s11658-012-0032-5. Epub 2012 Sep 20. Cell Mol Biol Lett. 2012. PMID: 23001513 Free PMC article. Review.
Cited by
-
Tumor suppression by resistant maltodextrin, Fibersol-2.Cancer Biol Ther. 2015;16(3):460-5. doi: 10.1080/15384047.2015.1009269. Cancer Biol Ther. 2015. PMID: 25692338 Free PMC article.
-
Corticospinal Motor Neurons Are Susceptible to Increased ER Stress and Display Profound Degeneration in the Absence of UCHL1 Function.Cereb Cortex. 2015 Nov;25(11):4259-72. doi: 10.1093/cercor/bhu318. Epub 2015 Jan 16. Cereb Cortex. 2015. PMID: 25596590 Free PMC article.
-
Anti-Oxidative, Anti-Inflammatory and Anti-Apoptotic Effects of Flavonols: Targeting Nrf2, NF-κB and p53 Pathways in Neurodegeneration.Antioxidants (Basel). 2021 Oct 15;10(10):1628. doi: 10.3390/antiox10101628. Antioxidants (Basel). 2021. PMID: 34679762 Free PMC article. Review.
-
Proteasome and Neuroprotective Effect of Hyperbaric Oxygen Preconditioning in Experimental Global Cerebral Ischemia in Rats.Front Neurol. 2022 Feb 17;13:812581. doi: 10.3389/fneur.2022.812581. eCollection 2022. Front Neurol. 2022. PMID: 35250819 Free PMC article.
-
Uncoupling of PUMA Expression and Apoptosis Contributes to Functional Heterogeneity in Renal Cell Carcinoma - Prognostic and Translational Implications.Transl Oncol. 2015 Dec;8(6):480-6. doi: 10.1016/j.tranon.2015.11.003. Transl Oncol. 2015. PMID: 26692529 Free PMC article.
References
-
- Anan, A., E. S. Baskin-Bey, S. F. Bronk, N. W. Werneburg, V. H. Shah, and G. J. Gores. 2006. Proteasome inhibition induces hepatic stellate cell apoptosis. Hepatology 43:335-344. - PubMed
-
- Berndtsson, M., M. Beaujouin, L. Rickardson, A. M. Havelka, R. Larsson, J. Westman, E. Liaudet-Coopman, and S. Linder. 2009. Induction of the lysosomal apoptosis pathway by inhibitors of the ubiquitin-proteasome system. Int. J. Cancer 124:1463-1469. - PubMed
-
- Biswas, S. C., E. Ryu, C. Park, C. Malagelada, and L. A. Greene. 2005. Puma and p53 play required roles in death evoked in a cellular model of Parkinson disease. Neurochem. Res. 30:839-845. - PubMed
-
- Bonfoco, E., S. Ceccatelli, L. Manzo, and P. Nicotera. 1995. Colchicine induces apoptosis in cerebellar granule cells. Exp. Cell Res. 218:189-200. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous