

Discovery of mutations in Saccharomyces cerevisiae by pooled linkage analysis and whole-genome sequencing
- PMID: 20923977
- PMCID: PMC2998298
- DOI: 10.1534/genetics.110.123232
Discovery of mutations in Saccharomyces cerevisiae by pooled linkage analysis and whole-genome sequencing
Abstract
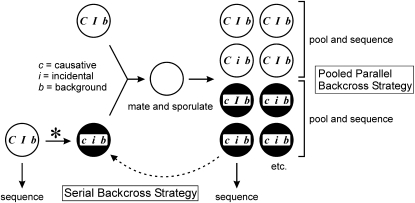
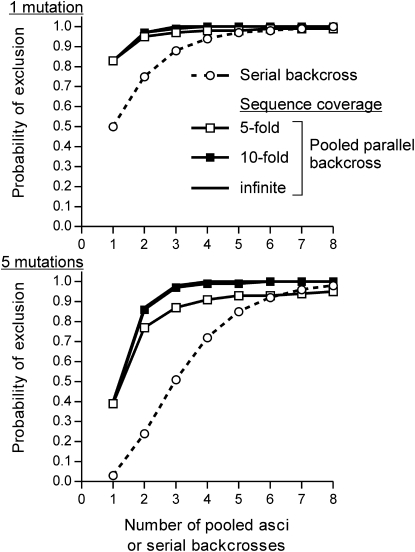
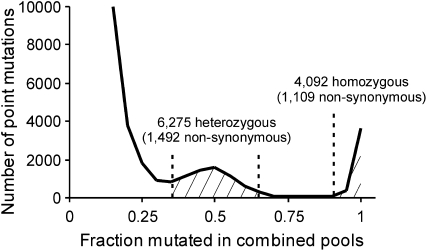
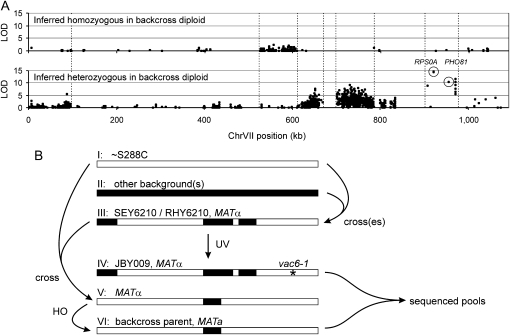
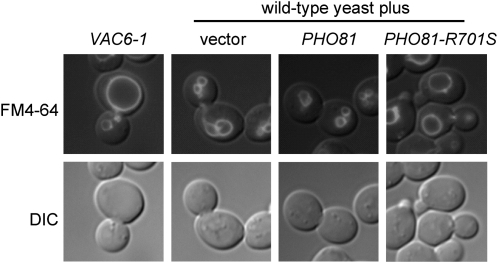
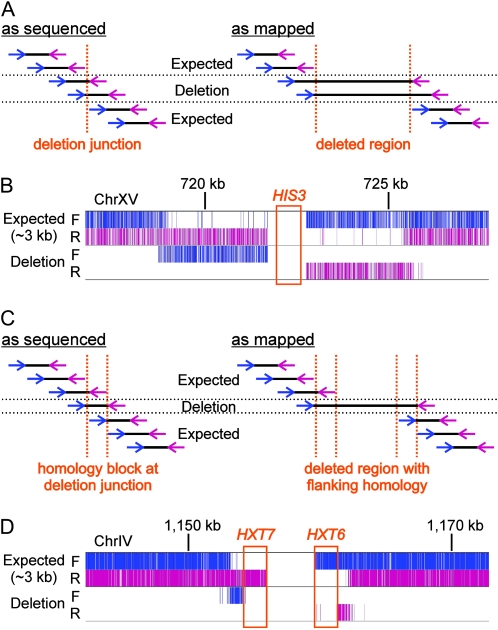
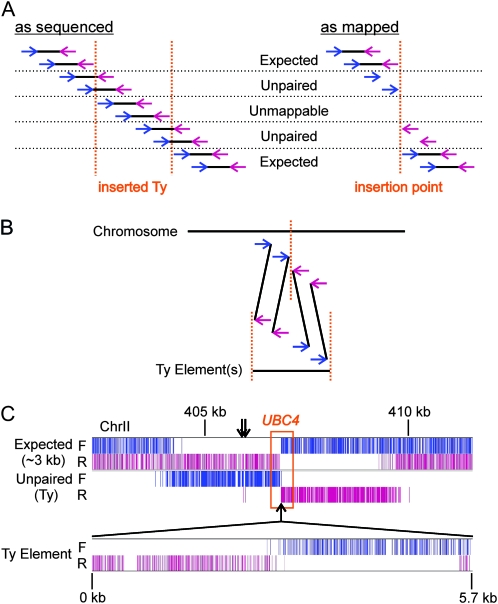
Many novel and important mutations arise in model organisms and human patients that can be difficult or impossible to identify using standard genetic approaches, especially for complex traits. Working with a previously uncharacterized dominant Saccharomyces cerevisiae mutant with impaired vacuole inheritance, we developed a pooled linkage strategy based on next-generation DNA sequencing to specifically identify functional mutations from among a large excess of polymorphisms, incidental mutations, and sequencing errors. The VAC6-1 mutation was verified to correspond to PHO81-R701S, the highest priority candidate reported by VAMP, the new software platform developed for these studies. Sequence data further revealed the large extent of strain background polymorphisms and structural alterations present in the host strain, which occurred by several mechanisms including a novel Ty insertion. The results provide a snapshot of the ongoing genomic changes that ultimately result in strain divergence and evolution, as well as a general model for the discovery of functional mutations in many organisms.
Figures
References
-
- Campagna, D., A. Albiero, A. Bilardi, E. Caniato, C. Forcato et al., 2009. PASS: a program to align short sequences. Bioinformatics 25 967–968. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
