

Coupling of receptor conformation and ligand orientation determine graded activity
- PMID: 20924370
- PMCID: PMC2974172
- DOI: 10.1038/nchembio.451
Coupling of receptor conformation and ligand orientation determine graded activity
Abstract
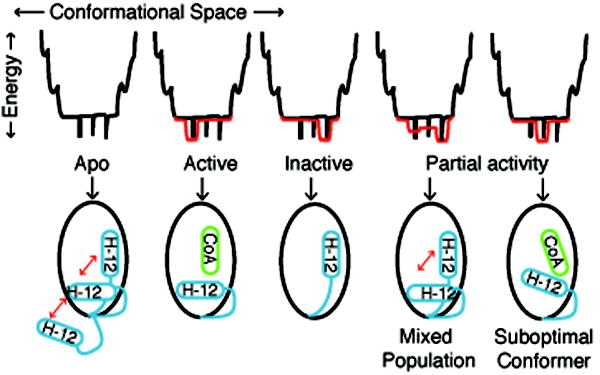
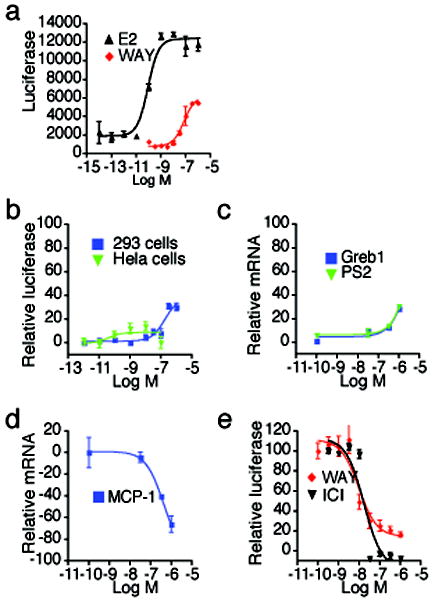
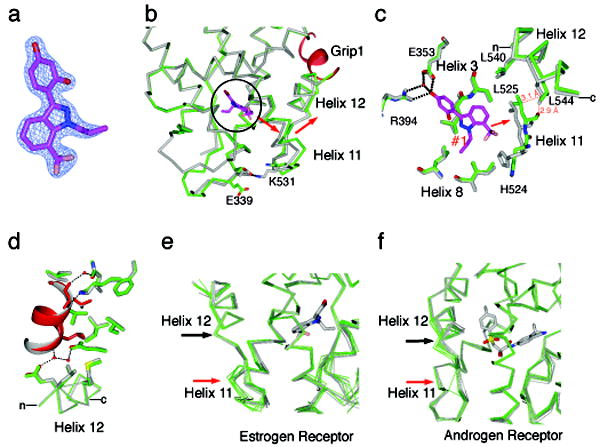
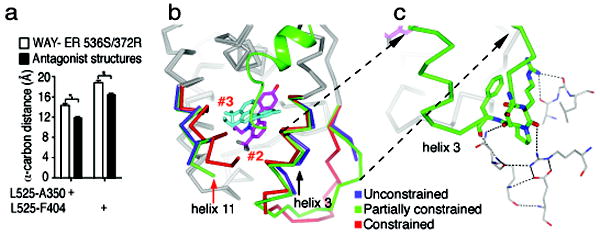
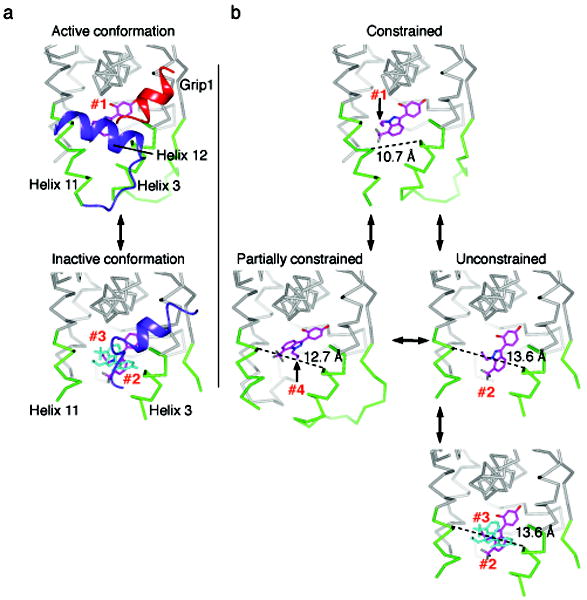
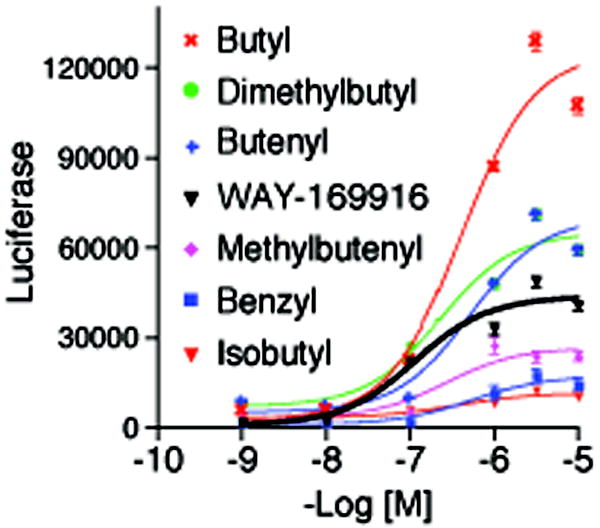
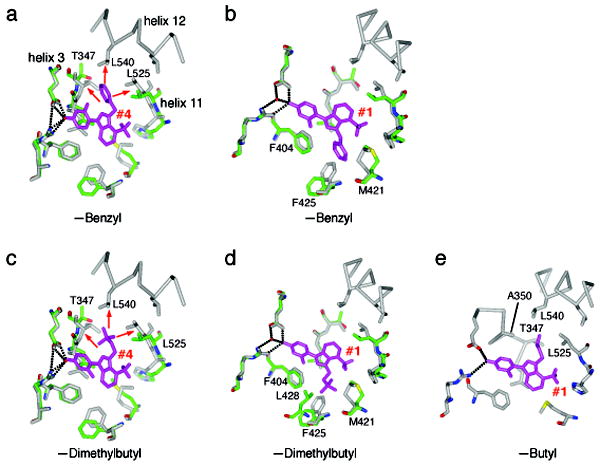
Small molecules stabilize specific protein conformations from a larger ensemble, enabling molecular switches that control diverse cellular functions. We show here that the converse also holds true: the conformational state of the estrogen receptor can direct distinct orientations of the bound ligand. 'Gain-of-allostery' mutations that mimic the effects of ligand in driving protein conformation allowed crystallization of the partial agonist ligand WAY-169916 with both the canonical active and inactive conformations of the estrogen receptor. The intermediate transcriptional activity induced by WAY-169916 is associated with the ligand binding differently to the active and inactive conformations of the receptor. Analyses of a series of chemical derivatives demonstrated that altering the ensemble of ligand binding orientations changes signaling output. The coupling of different ligand binding orientations to distinct active and inactive protein conformations defines a new mechanism for titrating allosteric signaling activity.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
