

Comprehensive genetic analysis of 182 unrelated families with congenital adrenal hyperplasia due to 21-hydroxylase deficiency
- PMID: 20926536
- PMCID: PMC3038490
- DOI: 10.1210/jc.2010-0319
Comprehensive genetic analysis of 182 unrelated families with congenital adrenal hyperplasia due to 21-hydroxylase deficiency
Abstract
Background: Genetic analysis is commonly performed in patients with congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency.
Study objective: The objective of the study was to describe comprehensive CYP21A2 mutation analysis in a large cohort of CAH patients.
Methods: Targeted CYP21A2 mutation analysis was performed in 213 patients and 232 parents from 182 unrelated families. Complete exons of CYP21A2 were sequenced in patients in whom positive mutations were not identified by targeted mutation analysis. Copy number variation and deletions were determined using Southern blot analysis and PCR methods. Genotype was correlated with phenotype.
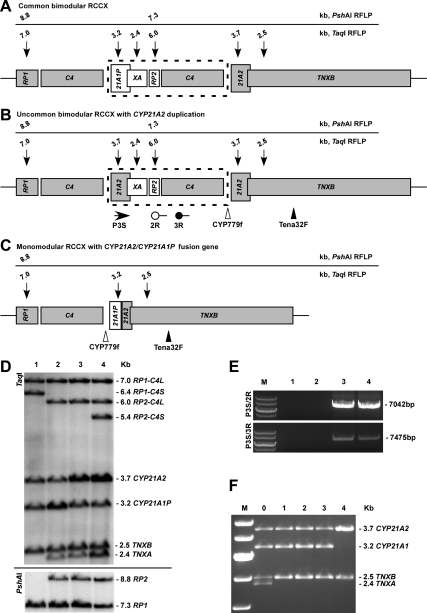
Results: In our heterogeneous U.S. cohort, targeted CYP21A2 mutation analysis did not identify mutations on one allele in 19 probands (10.4%). Sequencing identified six novel mutations (p.Gln262fs, IVS8+1G>A, IVS9-1G>A, p.R408H, p.Gly424fs, p.R426P) and nine previously reported rare mutations. The majority of patients (79%) were compound heterozygotes and 69% of nonclassic (NC) patients were compound heterozygous for a classic and a NC mutation. Duplicated CYP21A2 haplotypes, de novo mutations and uniparental disomy were present in 2.7% of probands and 1.9 and 0.9% of patients from informative families, respectively. Genotype accurately predicted phenotype in 90.5, 85.1, and 97.8% of patients with salt-wasting, simple virilizing, and NC mutations, respectively.
Conclusions: Extensive genetic analysis beyond targeted CYP21A2 mutational detection is often required to accurately determine genotype in patients with CAH due to the high frequency of complex genetic variation.
Figures
References
-
- Merke DP, Bornstein SR 2005 Congenital adrenal hyperplasia. Lancet 365:2125–2136 - PubMed
-
- Therrell Jr BL, Berenbaum SA, Manter-Kapanke V, Simmank J, Korman K, Prentice L, Gonzalez J, Gunn S 1998 Results of screening 1.9 million Texas newborns for 21-hydroxylase-deficient congenital adrenal hyperplasia. Pediatrics 101:583–590 - PubMed
-
- van der Kamp HJ, Wit JM 2004 Neonatal screening for congenital adrenal hyperplasia. Eur J Endocrinol 151(Suppl 3):U71–U75 - PubMed
-
- Fitness J, Dixit N, Webster D, Torresani T, Pergolizzi R, Speiser PW, Day DJ 1999 Genotyping of CYP21, linked chromosome 6p markers, and a sex-specific gene in neonatal screening for congenital adrenal hyperplasia. J Clin Endocrinol Metab 84:960–966 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
