

Metagenomic analysis of the viromes of three North American bat species: viral diversity among different bat species that share a common habitat
- PMID: 20926577
- PMCID: PMC3004358
- DOI: 10.1128/JVI.01255-10
Metagenomic analysis of the viromes of three North American bat species: viral diversity among different bat species that share a common habitat
Abstract
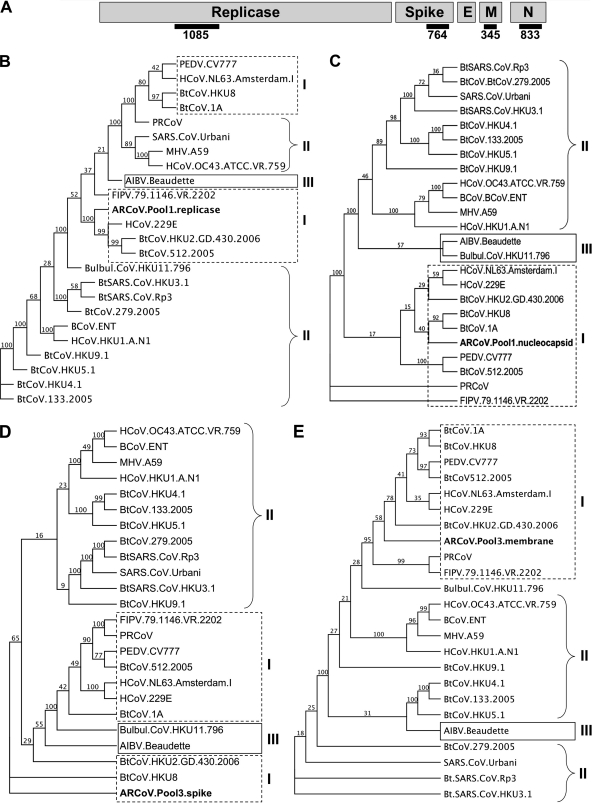
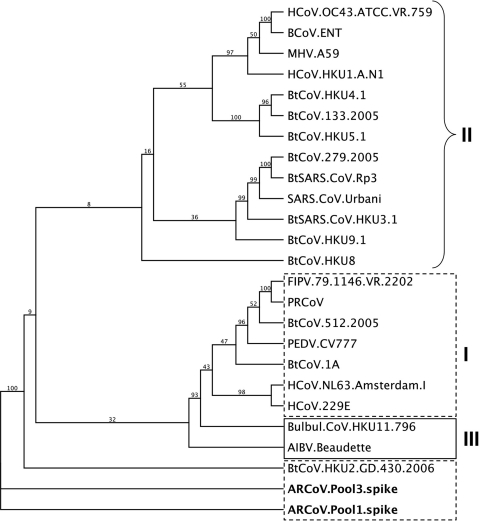
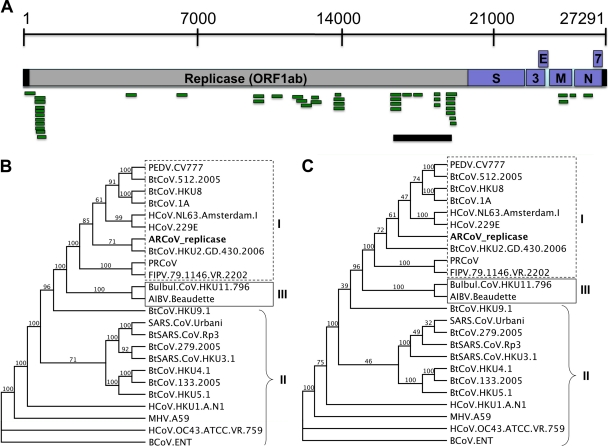
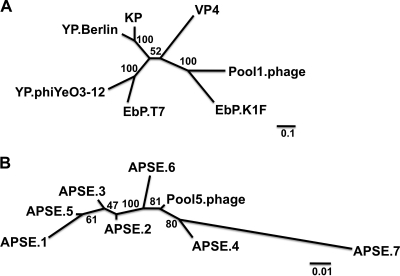
Effective prediction of future viral zoonoses requires an in-depth understanding of the heterologous viral population in key animal species that will likely serve as reservoir hosts or intermediates during the next viral epidemic. The importance of bats as natural hosts for several important viral zoonoses, including Ebola, Marburg, Nipah, Hendra, and rabies viruses and severe acute respiratory syndrome-coronavirus (SARS-CoV), has been established; however, the large viral population diversity (virome) of bats has been partially determined for only a few of the ∼1,200 bat species. To assess the virome of North American bats, we collected fecal, oral, urine, and tissue samples from individual bats captured at an abandoned railroad tunnel in Maryland that is cohabitated by 7 to 10 different bat species. Here, we present preliminary characterization of the virome of three common North American bat species, including big brown bats (Eptesicus fuscus), tricolored bats (Perimyotis subflavus), and little brown myotis (Myotis lucifugus). In samples derived from these bats, we identified viral sequences that were similar to at least three novel group 1 CoVs, large numbers of insect and plant virus sequences, and nearly full-length genomic sequences of two novel bacteriophages. These observations suggest that bats encounter and disseminate a large assortment of viruses capable of infecting many different animals, insects, and plants in nature.
Figures



References
-
- Arias, C. F., M. Escalera-Zamudio, L. Soto-Del Rio Mde, A. G. Cobian-Guemes, P. Isa, and S. Lopez. 2009. Molecular anatomy of 2009 influenza virus A (H1N1). Arch. Med. Res. 40:643-654. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
