

Role of epithelial HCO3⁻ transport in mucin secretion: lessons from cystic fibrosis
- PMID: 20926781
- PMCID: PMC3006319
- DOI: 10.1152/ajpcell.00362.2010
Role of epithelial HCO3⁻ transport in mucin secretion: lessons from cystic fibrosis
Abstract
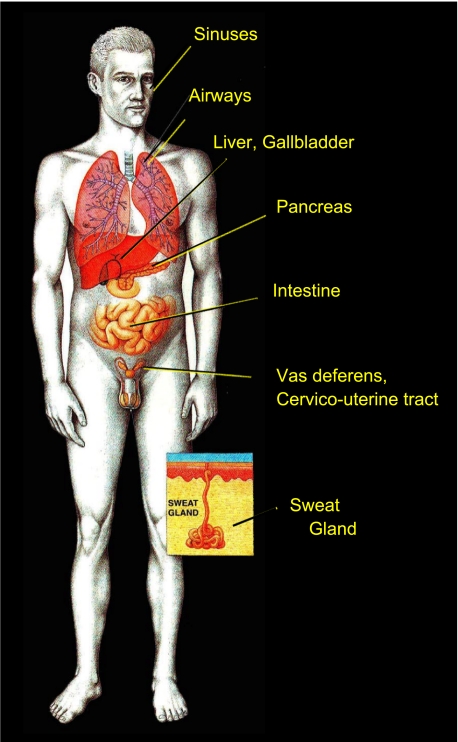
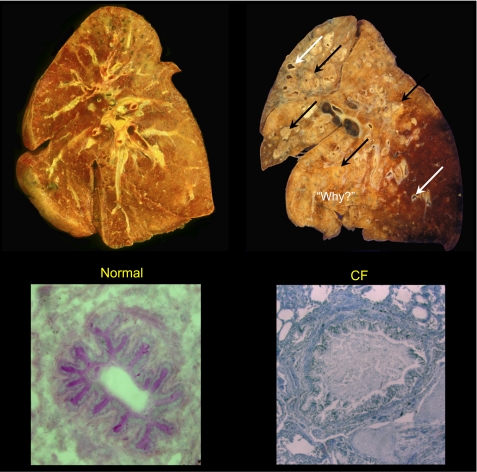
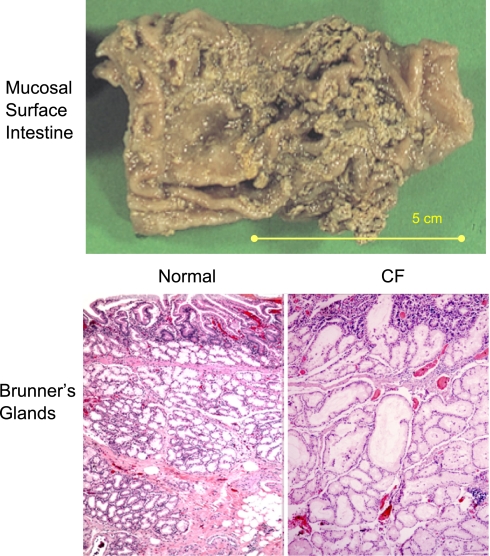
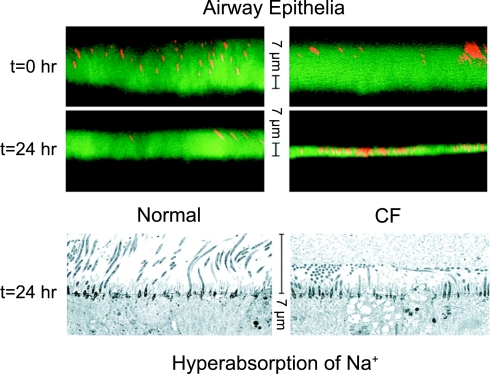
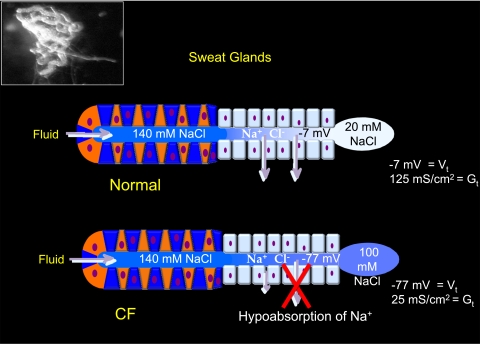
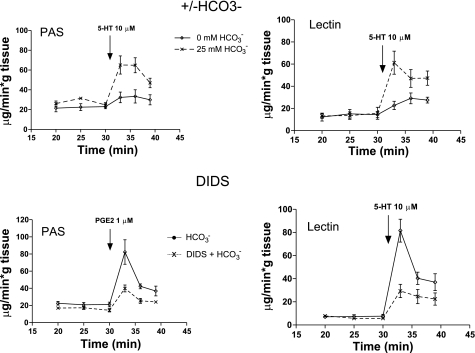
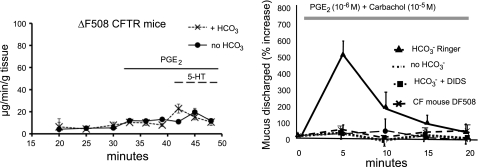
The invitation to present the 2010 Hans Ussing lecture for the Epithelial Transport Group of the American Physiological Society offered me a unique, special, and very surprising opportunity to join in saluting a man whom I met only once, but whose work was the basis, not only for my career, but also for finding the molecular defect in the inherited disease cystic fibrosis (CF). In this context, I will venture to make the tribute with a new explanation of why a mutation in a single gene that codes for an anion channel can cause devastation of multiple epithelial systems with pathogenic mucus. In so doing, I hope to raise awareness of a new role for that peculiar anion around which so much physiology revolves, HCO(3)(-). I begin by introducing CF pathology as I question the name of the disease as well as the prevalent view of the basis of its pathology by considering: 1) mucus, 2) salt, and 3) HCO(3)(-). I then present recent data showing that HCO(3)(-) is required for normal mucus discharge, and I will close with conjecture as to how HCO(3)(-) may support mucus discharge and why the failure to transport this electrolyte is pathogenic in CF.
Figures
References
-
- Aitken ML, Verdugo P. Donnan mechanism of mucin release and conditioning in goblet cells: the role of polyions. Symp Soc Exp Biol 43: 73–80, 1989 - PubMed
-
- Akiba Y, Guth PH, Engel E, Nastaskin I, Kaunitz JD. Dynamic regulation of mucus gel thickness in rat duodenum. Am J Physiol Gastrointest Liver Physiol 279: G437–G447, 2000 - PubMed
-
- Allen A, Hutton DA, Pearson JP. The MUC2 gene product: a human intestinal mucin. Int J Biochem Cell Biol 30: 797–801, 1998 - PubMed
-
- Andersen DH. Cystic fibrosis of the pancreas and its relation to celiac disease. Am J Dis Children 56: 344–399, 1938
-
- Bijman J, Quinton P. Permeability properties of cell membranes and tight junctions of normal and cystic fibrosis sweat ducts. Pflugers Arch 408: 505–510, 1987 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous