

Ontology-based meta-analysis of global collections of high-throughput public data
- PMID: 20927376
- PMCID: PMC2947508
- DOI: 10.1371/journal.pone.0013066
Ontology-based meta-analysis of global collections of high-throughput public data
Abstract
Background: The investigation of the interconnections between the molecular and genetic events that govern biological systems is essential if we are to understand the development of disease and design effective novel treatments. Microarray and next-generation sequencing technologies have the potential to provide this information. However, taking full advantage of these approaches requires that biological connections be made across large quantities of highly heterogeneous genomic datasets. Leveraging the increasingly huge quantities of genomic data in the public domain is fast becoming one of the key challenges in the research community today.
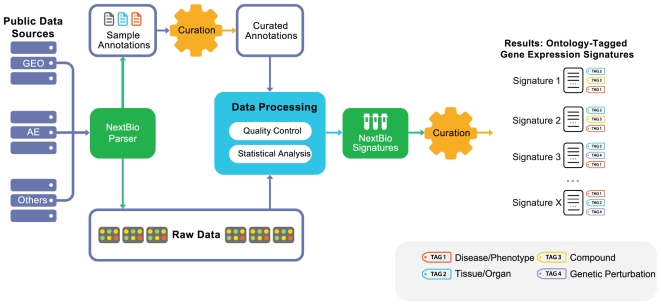
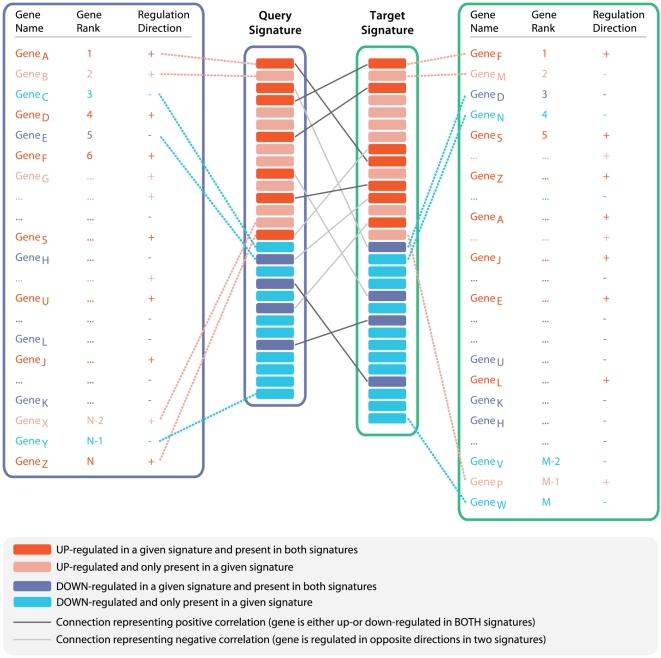
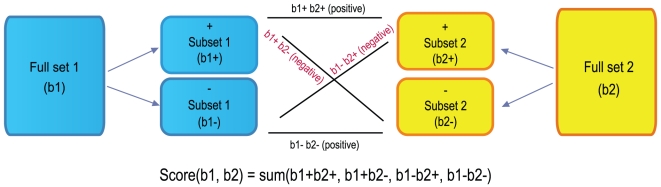
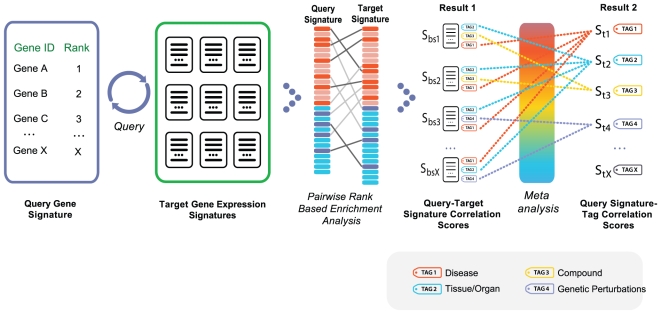
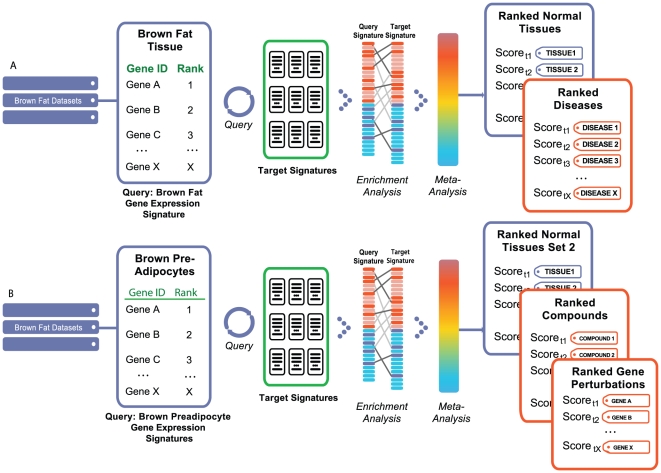
Methodology/results: We have developed a novel data mining framework that enables researchers to use this growing collection of public high-throughput data to investigate any set of genes or proteins. The connectivity between molecular states across thousands of heterogeneous datasets from microarrays and other genomic platforms is determined through a combination of rank-based enrichment statistics, meta-analyses, and biomedical ontologies. We address data quality concerns through dataset replication and meta-analysis and ensure that the majority of the findings are derived using multiple lines of evidence. As an example of our strategy and the utility of this framework, we apply our data mining approach to explore the biology of brown fat within the context of the thousands of publicly available gene expression datasets.
Conclusions: Our work presents a practical strategy for organizing, mining, and correlating global collections of large-scale genomic data to explore normal and disease biology. Using a hypothesis-free approach, we demonstrate how a data-driven analysis across very large collections of genomic data can reveal novel discoveries and evidence to support existing hypothesis.
Conflict of interest statement
Figures
References
-
- Gardiner-Garden M, Littlejohn TG. A comparison of microarray databases. Brief Bioinform. 2001;2:143–158. - PubMed
-
- Rhodes DR, Barrette TR, Rubin MA, Ghosh D, Chinnaiyan AM. Meta-analysis of microarrays: interstudy validation of gene expression profiles reveals pathway dysregulation in prostate cancer. Cancer Res. 2002;62:4427–4433. - PubMed
-
- Ghosh D, Barette TR, Rhodes D, Chinnaiyan AM. Statistical issues and methods for meta-analysis of microarray data: a case study in prostate cancer. Funct Integr Genomics. 2003;3:180–188. - PubMed
-
- Griffith OL, Melck A, Jones SJM, Wiseman SM. Meta-analysis and meta-review of thyroid cancer gene expression profiling studies identifies important diagnostic biomarkers. J Clin Oncol. 2006;24:5043–5051. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
