

Mutations that permit residual CFTR function delay acquisition of multiple respiratory pathogens in CF patients
- PMID: 20932301
- PMCID: PMC2964615
- DOI: 10.1186/1465-9921-11-140
Mutations that permit residual CFTR function delay acquisition of multiple respiratory pathogens in CF patients
Abstract
Background: Lung infection by various organisms is a characteristic feature of cystic fibrosis (CF). CFTR genotype effects acquisition of Pseudomonas aeruginosa (Pa), however the effect on acquisition of other infectious organisms that frequently precede Pa is relatively unknown. Understanding the role of CFTR in the acquisition of organisms first detected in patients may help guide symptomatic and molecular-based treatment for CF.
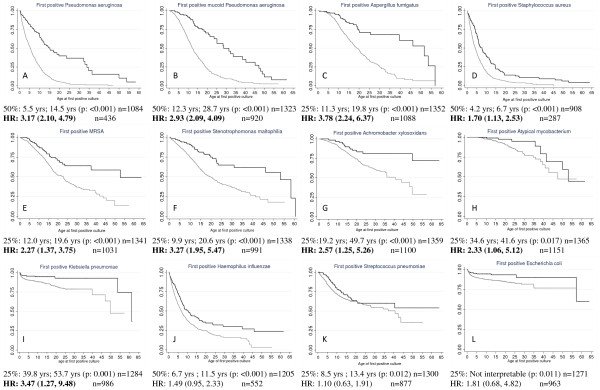
Methods: Lung infection, defined as a single positive respiratory tract culture, was assessed for 13 organisms in 1,381 individuals with CF. Subjects were divided by predicted CFTR function: 'Residual': carrying at least one partial function CFTR mutation (class IV or V) and 'Minimal' those who do not carry a partial function mutation. Kaplan-Meier estimates were created to assess CFTR effect on age of acquisition for each organism. Cox proportional hazard models were performed to control for possible cofactors. A separate Cox regression was used to determine whether defining infection with Pa, mucoid Pa or Aspergillus (Asp) using alternative criteria affected the results. The influence of severity of lung disease at the time of acquisition was evaluated using stratified Cox regression methods by lung disease categories.
Results: Subjects with 'Minimal' CFTR function had a higher hazard than patients with 'Residual' function for acquisition of 9 of 13 organisms studied (HR ranging from 1.7 to 3.78 based on the organism studied). Subjects with minimal CFTR function acquired infection at a younger age than those with residual function for 12 of 13 organisms (p-values ranging: < 0.001 to 0.017). Minimal CFTR function also associated with younger age of infection when 3 alternative definitions of infection with Pa, mucoid Pa or Asp were employed. Risk of infection is correlated with CFTR function for 8 of 9 organisms in patients with good lung function (>90%ile) but only 1 of 9 organisms in those with poorer lung function (<50%ile).
Conclusions: Residual CFTR function correlates with later onset of respiratory tract infection by a wide spectrum of organisms frequently cultured from CF patients. The protective effect conferred by residual CFTR function is diminished in CF patients with more advanced lung disease.
Figures
Similar articles
-
CFTR genotype and clinical outcomes of adult patients carried as cystic fibrosis disease.Gene. 2014 May 1;540(2):183-90. doi: 10.1016/j.gene.2014.02.040. Epub 2014 Feb 26. Gene. 2014. PMID: 24583165
-
CFTR Cl- channel function in native human colon correlates with the genotype and phenotype in cystic fibrosis.Gastroenterology. 2004 Oct;127(4):1085-95. doi: 10.1053/j.gastro.2004.07.006. Gastroenterology. 2004. PMID: 15480987
-
Cystic fibrosis transmembrane conductance regulator (CFTR)-mediated residual chloride secretion does not protect against early chronic Pseudomonas aeruginosa infection in F508del homozygous cystic fibrosis patients.Pediatr Res. 2004 Jan;55(1):69-75. doi: 10.1203/01.PDR.0000100758.66805.CE. Epub 2003 Nov 6. Pediatr Res. 2004. PMID: 14605249
-
CFTR mutations and host susceptibility to Pseudomonas aeruginosa lung infection.Curr Opin Microbiol. 2002 Feb;5(1):81-6. doi: 10.1016/s1369-5274(02)00290-4. Curr Opin Microbiol. 2002. PMID: 11834374 Review.
-
Mouse models of chronic lung infection with Pseudomonas aeruginosa: models for the study of cystic fibrosis.Pediatr Pulmonol. 2000 Nov;30(5):413-24. doi: 10.1002/1099-0496(200011)30:5<413::aid-ppul8>3.0.co;2-9. Pediatr Pulmonol. 2000. PMID: 11064433 Review.
Cited by
-
Association of lung function, chest radiographs and clinical features in infants with cystic fibrosis.Eur Respir J. 2013 Dec;42(6):1545-52. doi: 10.1183/09031936.00138412. Epub 2013 May 30. Eur Respir J. 2013. PMID: 23722613 Free PMC article.
-
CFTR function and clinical response to modulators parallel nasal epithelial organoid swelling.Am J Physiol Lung Cell Mol Physiol. 2021 Jul 1;321(1):L119-L129. doi: 10.1152/ajplung.00639.2020. Epub 2021 May 19. Am J Physiol Lung Cell Mol Physiol. 2021. PMID: 34009038 Free PMC article.
-
Chloride Conductance, Nasal Potential Difference and Cystic Fibrosis Pathophysiology.Lung. 2020 Feb;198(1):151-156. doi: 10.1007/s00408-019-00293-6. Epub 2019 Nov 16. Lung. 2020. PMID: 31734731
-
Functional variants in the cystic fibrosis transmembrane conductance regulator (CFTR) gene are associated with increased risk of colorectal cancer.Hum Mol Genet. 2025 Mar 20;34(7):617-625. doi: 10.1093/hmg/ddaf007. Hum Mol Genet. 2025. PMID: 39825500
-
Mutations of the cystic fibrosis transmembrane conductance regulator gene in males with congenital bilateral absence of the vas deferens: Reproductive implications and genetic counseling (Review).Mol Med Rep. 2020 Nov;22(5):3587-3596. doi: 10.3892/mmr.2020.11456. Epub 2020 Aug 24. Mol Med Rep. 2020. PMID: 33000223 Free PMC article. Review.
References
-
- Halliburton CS, Mannino DM, Olney RS. Cystic fibrosis deaths in the United States from 1979 through 1991. An analysis using multiple-cause mortality data. Arch Pediatr Adolesc Med. 1996;150:1181–1185. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
