

Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors
- PMID: 20937875
- PMCID: PMC2973891
- DOI: 10.1073/pnas.1007028107
Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors
Abstract
In vertebrates, including humans, individuals harbor gut microbial communities whose species composition and relative proportions of dominant microbial groups are tremendously varied. Although external and stochastic factors clearly contribute to the individuality of the microbiota, the fundamental principles dictating how environmental factors and host genetic factors combine to shape this complex ecosystem are largely unknown and require systematic study. Here we examined factors that affect microbiota composition in a large (n = 645) mouse advanced intercross line originating from a cross between C57BL/6J and an ICR-derived outbred line (HR). Quantitative pyrosequencing of the microbiota defined a core measurable microbiota (CMM) of 64 conserved taxonomic groups that varied quantitatively across most animals in the population. Although some of this variation can be explained by litter and cohort effects, individual host genotype had a measurable contribution. Testing of the CMM abundances for cosegregation with 530 fully informative SNP markers identified 18 host quantitative trait loci (QTL) that show significant or suggestive genome-wide linkage with relative abundances of specific microbial taxa. These QTL affect microbiota composition in three ways; some loci control individual microbial species, some control groups of related taxa, and some have putative pleiotropic effects on groups of distantly related organisms. These data provide clear evidence for the importance of host genetic control in shaping individual microbiome diversity in mammals, a key step toward understanding the factors that govern the assemblages of gut microbiota associated with complex diseases.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
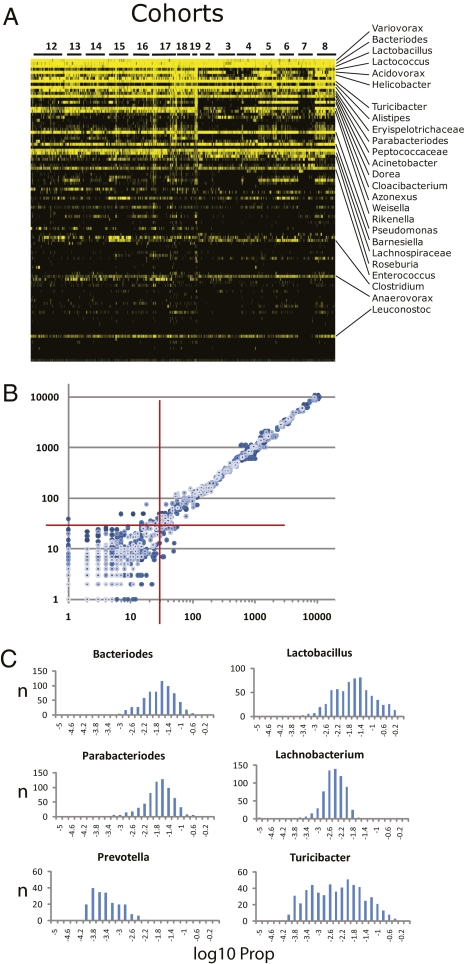
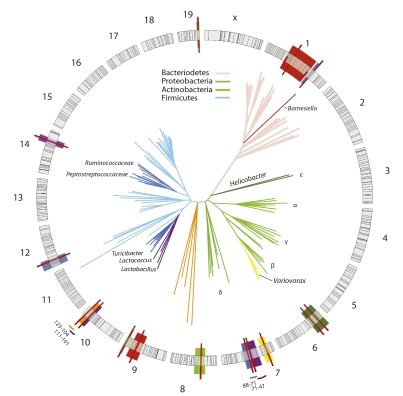
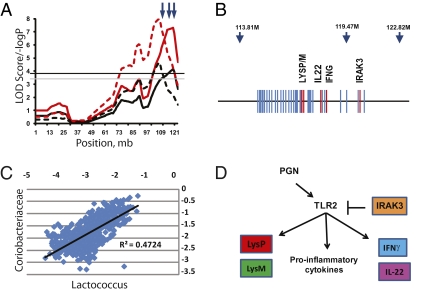
Comment in
-
Host genes shape the gut microbiota.Nat Rev Microbiol. 2010 Dec;8(12):838. doi: 10.1038/nrmicro2479. Nat Rev Microbiol. 2010. PMID: 21125700 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
