

Geoseq: a tool for dissecting deep-sequencing datasets
- PMID: 20939882
- PMCID: PMC2972303
- DOI: 10.1186/1471-2105-11-506
Geoseq: a tool for dissecting deep-sequencing datasets
Abstract
Background: Datasets generated on deep-sequencing platforms have been deposited in various public repositories such as the Gene Expression Omnibus (GEO), Sequence Read Archive (SRA) hosted by the NCBI, or the DNA Data Bank of Japan (ddbj). Despite being rich data sources, they have not been used much due to the difficulty in locating and analyzing datasets of interest.
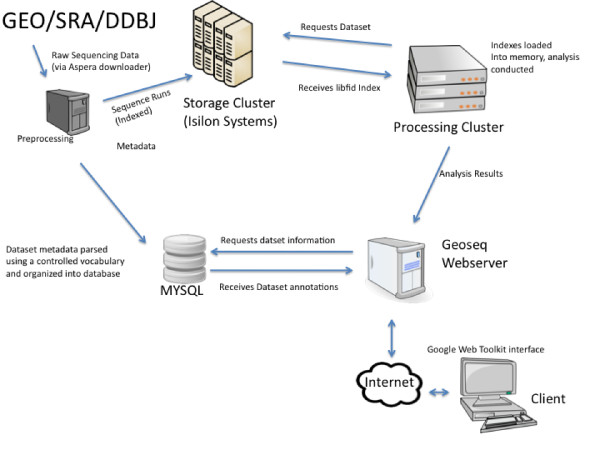
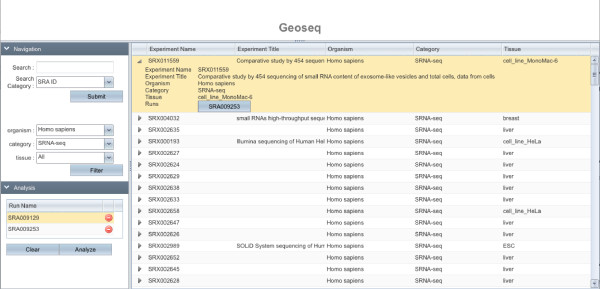
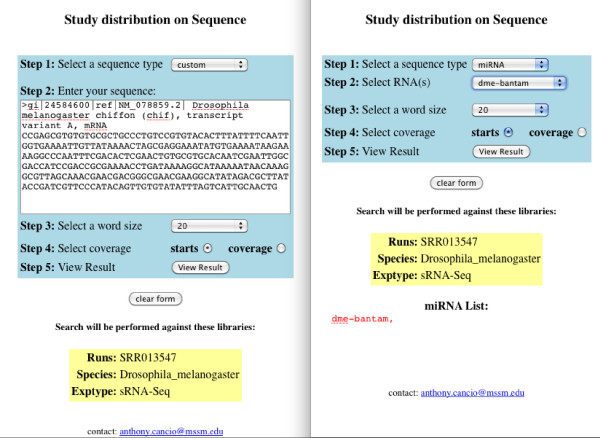
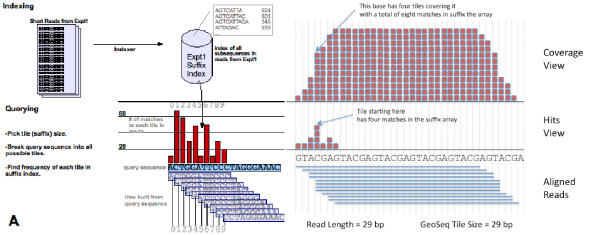
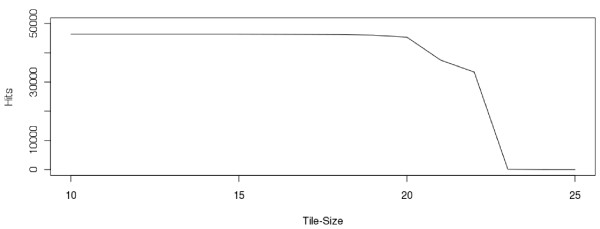
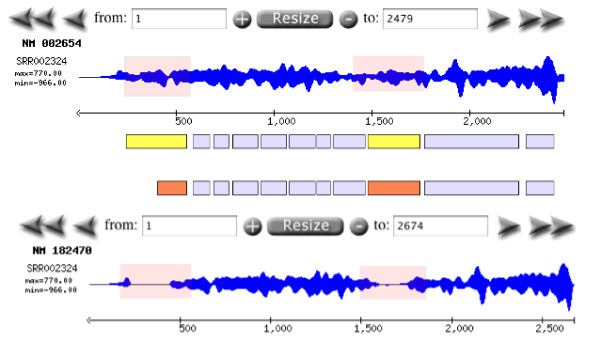
Results: Geoseq http://geoseq.mssm.edu provides a new method of analyzing short reads from deep sequencing experiments. Instead of mapping the reads to reference genomes or sequences, Geoseq maps a reference sequence against the sequencing data. It is web-based, and holds pre-computed data from public libraries. The analysis reduces the input sequence to tiles and measures the coverage of each tile in a sequence library through the use of suffix arrays. The user can upload custom target sequences or use gene/miRNA names for the search and get back results as plots and spreadsheet files. Geoseq organizes the public sequencing data using a controlled vocabulary, allowing identification of relevant libraries by organism, tissue and type of experiment.
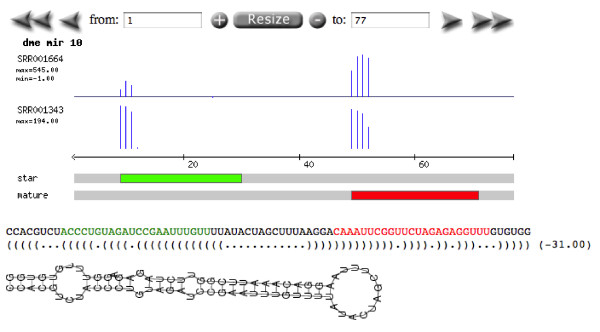
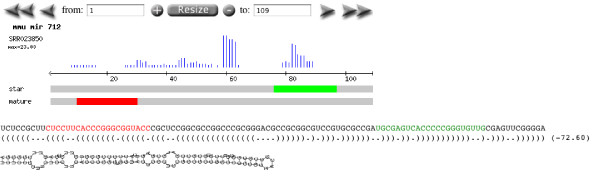
Conclusions: Analysis of small sets of sequences against deep-sequencing datasets, as well as identification of public datasets of interest, is simplified by Geoseq. We applied Geoseq to, a) identify differential isoform expression in mRNA-seq datasets, b) identify miRNAs (microRNAs) in libraries, and identify mature and star sequences in miRNAS and c) to identify potentially mis-annotated miRNAs. The ease of using Geoseq for these analyses suggests its utility and uniqueness as an analysis tool.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
