

GenHtr: a tool for comparative assessment of genetic heterogeneity in microbial genomes generated by massive short-read sequencing
- PMID: 20939910
- PMCID: PMC2967562
- DOI: 10.1186/1471-2105-11-508
GenHtr: a tool for comparative assessment of genetic heterogeneity in microbial genomes generated by massive short-read sequencing
Abstract
Background: Microevolution is the study of short-term changes of alleles within a population and their effects on the phenotype of organisms. The result of the below-species-level evolution is heterogeneity, where populations consist of subpopulations with a large number of structural variations. Heterogeneity analysis is thus essential to our understanding of how selective and neutral forces shape bacterial populations over a short period of time. The Solexa Genome Analyzer, a next-generation sequencing platform, allows millions of short sequencing reads to be obtained with great accuracy, allowing for the ability to study the dynamics of the bacterial population at the whole genome level. The tool referred to as GenHtr was developed for genome-wide heterogeneity analysis.
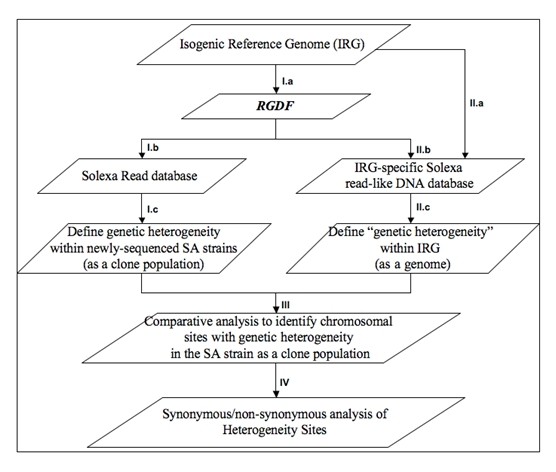
Results: For particular bacterial strains, GenHtr relies on a set of Solexa short reads on given bacteria pathogens and their isogenic reference genome to identify heterogeneity sites, the chromosomal positions with multiple variants of genes in the bacterial population, and variations that occur in large gene families. GenHtr accomplishes this by building and comparatively analyzing genome-wide heterogeneity genotypes for both the newly sequenced genomes (using massive short-read sequencing) and their isogenic reference (using simulated data). As proof of the concept, this approach was applied to SRX007711, the Solexa sequencing data for a newly sequenced Staphylococcus aureus subsp. USA300 cell line, and demonstrated that it could predict such multiple variants. They include multiple variants of genes critical in pathogenesis, e.g. genes encoding a LysR family transcriptional regulator, 23 S ribosomal RNA, and DNA mismatch repair protein MutS. The heterogeneity results in non-synonymous and nonsense mutations, leading to truncated proteins for both LysR and MutS.
Conclusion: GenHtr was developed for genome-wide heterogeneity analysis. Although it is much more time-consuming when compared to Maq, a popular tool for SNP analysis, GenHtr is able to predict potential multiple variants that pre-exist in the bacterial population as well as SNPs that occur in the highly duplicated gene families. It is expected that, with the proper experimental design, this analysis can improve our understanding of the molecular mechanism underlying the dynamics and the evolution of drug-resistant bacterial pathogens.
Figures
Similar articles
-
Gnom(Cmp): a quantitative approach for comparative analysis of closely related genomes of bacterial pathogens.Genome. 2011 May;54(5):402-18. doi: 10.1139/g11-005. Epub 2011 May 3. Genome. 2011. PMID: 21539441
-
Annotation-based genome-wide SNP discovery in the large and complex Aegilops tauschii genome using next-generation sequencing without a reference genome sequence.BMC Genomics. 2011 Jan 25;12:59. doi: 10.1186/1471-2164-12-59. BMC Genomics. 2011. PMID: 21266061 Free PMC article.
-
Using quality scores and longer reads improves accuracy of Solexa read mapping.BMC Bioinformatics. 2008 Feb 28;9:128. doi: 10.1186/1471-2105-9-128. BMC Bioinformatics. 2008. PMID: 18307793 Free PMC article.
-
NGSPanPipe: A Pipeline for Pan-genome Identification in Microbial Strains from Experimental Reads.Adv Exp Med Biol. 2018;1052:39-49. doi: 10.1007/978-981-10-7572-8_4. Adv Exp Med Biol. 2018. PMID: 29785479 Review.
-
Model-based quality assessment and base-calling for second-generation sequencing data.Biometrics. 2010 Sep;66(3):665-74. doi: 10.1111/j.1541-0420.2009.01353.x. Biometrics. 2010. PMID: 19912177 Free PMC article. Review.
Cited by
-
Evolution in fast forward: a potential role for mutators in accelerating Staphylococcus aureus pathoadaptation.J Bacteriol. 2013 Feb;195(3):615-28. doi: 10.1128/JB.00733-12. Epub 2012 Nov 30. J Bacteriol. 2013. PMID: 23204459 Free PMC article.
-
A Retrospective Study on Genetic Heterogeneity within Treponema Strains: Subpopulations Are Genetically Distinct in a Limited Number of Positions.PLoS Negl Trop Dis. 2015 Oct 5;9(10):e0004110. doi: 10.1371/journal.pntd.0004110. eCollection 2015. PLoS Negl Trop Dis. 2015. PMID: 26436423 Free PMC article.
References
-
- Feil EJ, Maiden MC, Achtman M, Spratt BG. The relative contributions of recombination and mutation to the divergence of clones of Neisseria meningitidis. Mol Biol Evol. 1999;16:1496–1502. - PubMed
-
- Maynard Smith J, Smith NH. Detecting recombination from gene trees. Mol Biol Evol. 1998;15:590–599. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
