

Neuronal gap junctions are required for NMDA receptor-mediated excitotoxicity: implications in ischemic stroke
- PMID: 20943940
- PMCID: PMC3007655
- DOI: 10.1152/jn.00656.2010
Neuronal gap junctions are required for NMDA receptor-mediated excitotoxicity: implications in ischemic stroke
Abstract
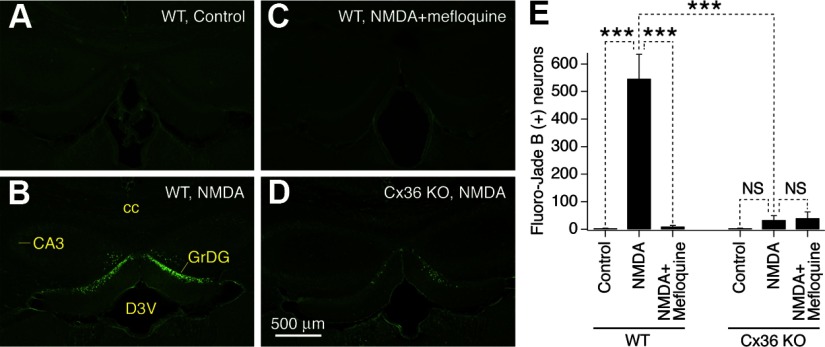
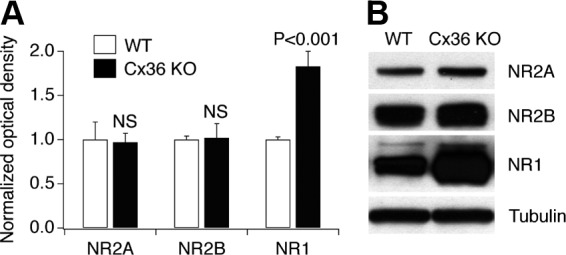
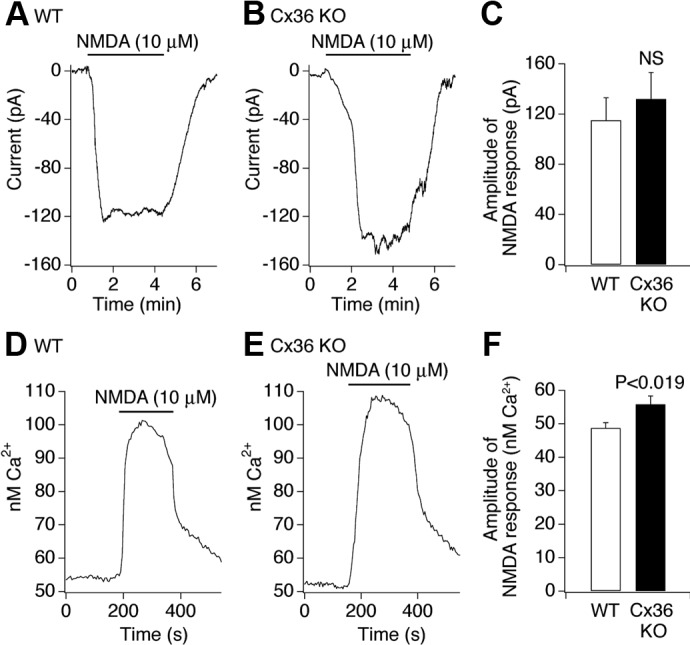
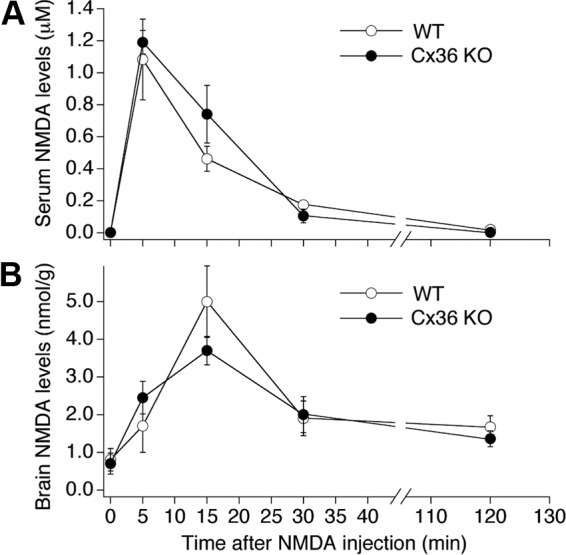
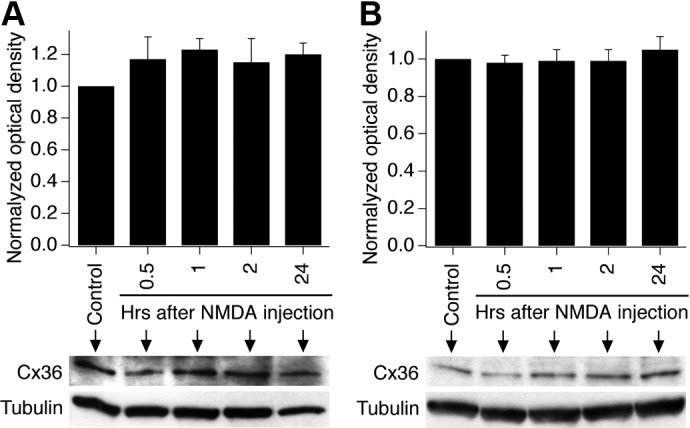
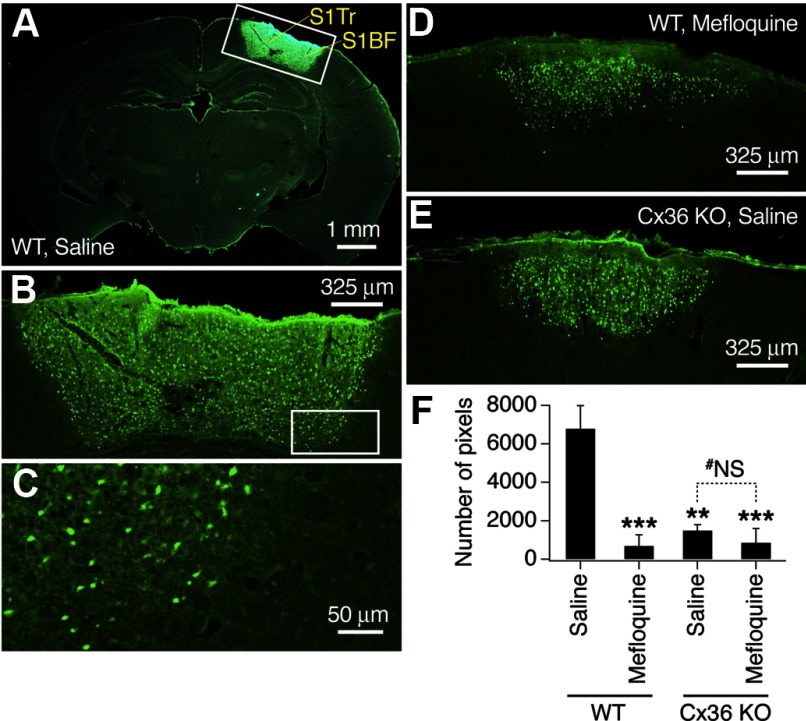
N-methyl-D-aspartate receptors (NMDARs) play an important role in cell survival versus cell death decisions during neuronal development, ischemia, trauma, and epilepsy. Coupling of neurons by electrical synapses (gap junctions) is high or increases in neuronal networks during all these conditions. In the developing CNS, neuronal gap junctions are critical for two different types of NMDAR-dependent cell death. However, whether neuronal gap junctions play a role in NMDAR-dependent neuronal death in the mature CNS was not known. Using Fluoro-Jade B staining, we show that a single intraperitoneal administration of NMDA (100 mg/kg) to adult wild-type mice induces neurodegeneration in three forebrain regions, including rostral dentate gyrus. However, the NMDAR-mediated neuronal death is prevented by pharmacological blockade of neuronal gap junctions (with mefloquine, 30 mg/kg) and does not occur in mice lacking neuronal gap junction protein, connexin 36. Using Western blots, electrophysiology, calcium imaging, and gas chromatography-mass spectrometry in wild-type and connexin 36 knockout mice, we show that the reduced level of neuronal death in knockout animals is not caused by the reduced expression of NMDARs, activity of NMDARs, or permeability of the blood-brain barrier to NMDA. In wild-type animals, this neuronal death is not caused by upregulation of connexin 36 by NMDA. Finally, pharmacological and genetic inactivation of neuronal gap junctions in mice also dramatically reduces neuronal death caused by photothrombotic focal cerebral ischemia. The results indicate that neuronal gap junctions are required for NMDAR-dependent excitotoxicity and play a critical role in ischemic neuronal death.
Figures
Similar articles
-
Gap junctions are required for NMDA receptor dependent cell death in developing neurons.J Neurophysiol. 2007 Nov;98(5):2878-86. doi: 10.1152/jn.00362.2007. Epub 2007 Sep 12. J Neurophysiol. 2007. PMID: 17855590
-
Death of Neurons following Injury Requires Conductive Neuronal Gap Junction Channels but Not a Specific Connexin.PLoS One. 2015 May 27;10(5):e0125395. doi: 10.1371/journal.pone.0125395. eCollection 2015. PLoS One. 2015. PMID: 26017008 Free PMC article.
-
Neuronal gap junctions play a role in the secondary neuronal death following controlled cortical impact.Neurosci Lett. 2012 Aug 22;524(1):16-9. doi: 10.1016/j.neulet.2012.06.065. Epub 2012 Jul 7. Neurosci Lett. 2012. PMID: 22781494 Free PMC article.
-
Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: A perspective on the contributions of apoptosis and necrosis.Brain Res Bull. 1998 Jul 1;46(4):281-309. doi: 10.1016/s0361-9230(98)00024-0. Brain Res Bull. 1998. PMID: 9671259 Review.
-
Novel model for the mechanisms of glutamate-dependent excitotoxicity: role of neuronal gap junctions.Brain Res. 2012 Dec 3;1487:123-30. doi: 10.1016/j.brainres.2012.05.063. Epub 2012 Jul 5. Brain Res. 2012. PMID: 22771704 Free PMC article. Review.
Cited by
-
Anticonvulsant effects of mefloquine on generalized tonic-clonic seizures induced by two acute models in rats.BMC Neurosci. 2015 Mar 1;16:7. doi: 10.1186/s12868-015-0145-7. BMC Neurosci. 2015. PMID: 25886955 Free PMC article.
-
Downregulation of Neuronal and Dendritic Connexin36-Made Electrical Synapses Without Glutamatergic Axon Terminals in Spinal Anterior Horn Cells From the Early Stage of Amyotrophic Lateral Sclerosis.Front Neurosci. 2018 Nov 28;12:894. doi: 10.3389/fnins.2018.00894. eCollection 2018. Front Neurosci. 2018. PMID: 30546295 Free PMC article.
-
A potential role for neuronal connexin 36 in the pathogenesis of amyotrophic lateral sclerosis.Neurosci Lett. 2018 Feb 14;666:1-4. doi: 10.1016/j.neulet.2017.12.027. Epub 2017 Dec 12. Neurosci Lett. 2018. PMID: 29246791 Free PMC article.
-
Deletion of neuronal gap junction protein connexin 36 impairs hippocampal LTP.Neurosci Lett. 2011 Sep 8;502(1):30-2. doi: 10.1016/j.neulet.2011.07.018. Epub 2011 Jul 20. Neurosci Lett. 2011. PMID: 21798314 Free PMC article.
-
Targeting autophagy in ischemic stroke: From molecular mechanisms to clinical therapeutics.Pharmacol Ther. 2021 Sep;225:107848. doi: 10.1016/j.pharmthera.2021.107848. Epub 2021 Apr 3. Pharmacol Ther. 2021. PMID: 33823204 Free PMC article. Review.
References
-
- Arumugam H, Liu X, Colombo PJ, Corriveau RA, Belousov AB. NMDA receptors regulate developmental gap junction uncoupling via CREB signaling. Nat Neurosci 8: 1720–1726, 2005 - PubMed
-
- Belluardo N, Mudo G, Trovato-Salinaro A, Le Gurun S, Charollais A, Serre-Beinier V, Amato G, Haefliger JA, Meda P, Condorelli DF. Expression of connexin36 in the adult and developing rat brain. Brain Res 865: 121–138, 2000 - PubMed
-
- Bennett MV, Zukin RS. Electrical coupling and neuronal synchronization in the Mammalian brain. Neuron 41: 495–511, 2004 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous