

Discovering gene functional relationships using FAUN (Feature Annotation Using Nonnegative matrix factorization)
- PMID: 20946597
- PMCID: PMC3026361
- DOI: 10.1186/1471-2105-11-S6-S14
Discovering gene functional relationships using FAUN (Feature Annotation Using Nonnegative matrix factorization)
Abstract
Background: Searching the enormous amount of information available in biomedical literature to extract novel functional relationships among genes remains a challenge in the field of bioinformatics. While numerous (software) tools have been developed to extract and identify gene relationships from biological databases, few effectively deal with extracting new (or implied) gene relationships, a process which is useful in interpretation of discovery-oriented genome-wide experiments.
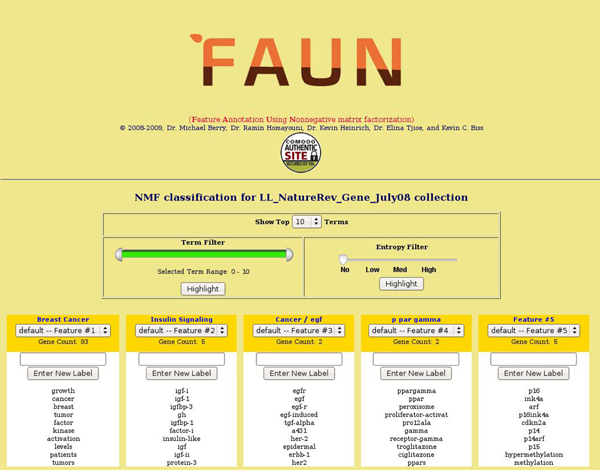
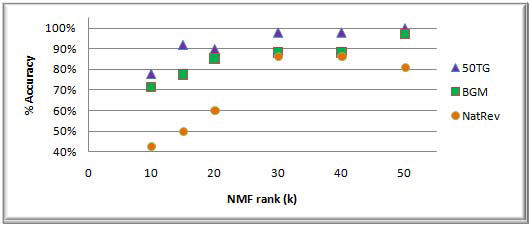
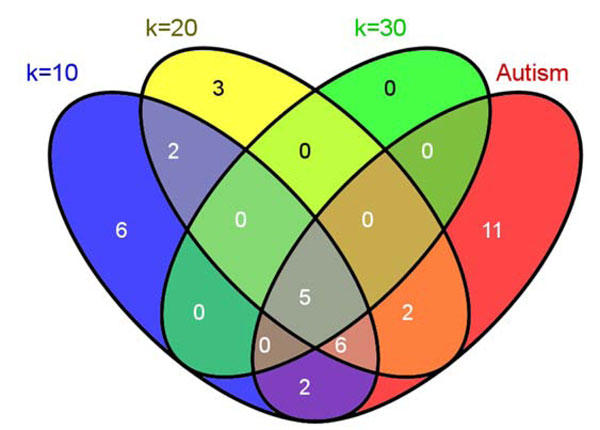
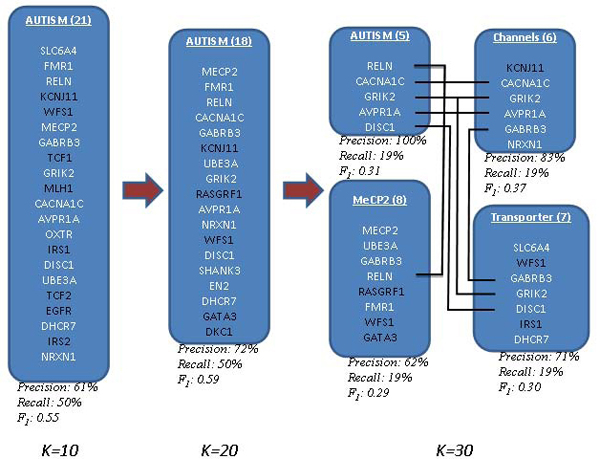
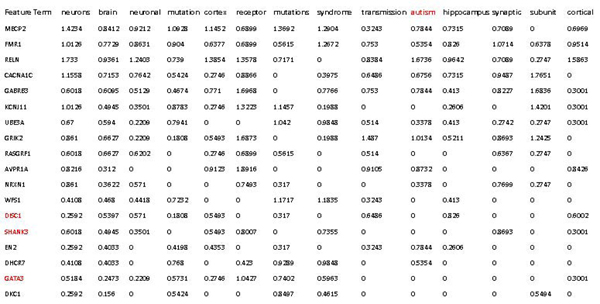
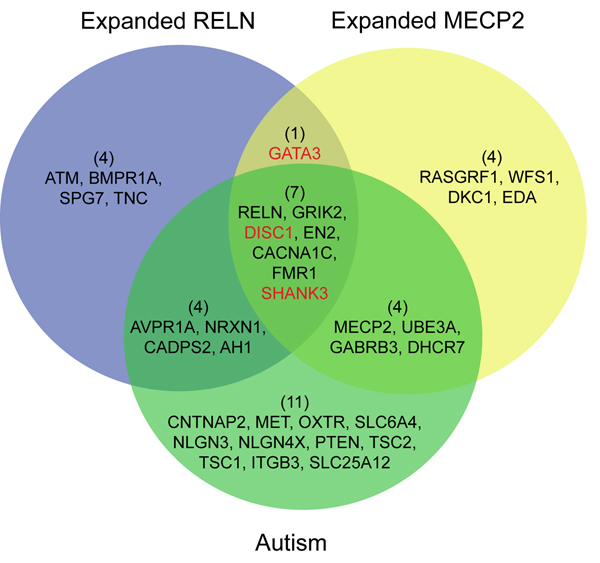
Results: In this study, we develop a Web-based bioinformatics software environment called FAUN or Feature Annotation Using Nonnegative matrix factorization (NMF) to facilitate both the discovery and classification of functional relationships among genes. Both the computational complexity and parameterization of NMF for processing gene sets are discussed. FAUN is tested on three manually constructed gene document collections. Its utility and performance as a knowledge discovery tool is demonstrated using a set of genes associated with Autism.
Conclusions: FAUN not only assists researchers to use biomedical literature efficiently, but also provides utilities for knowledge discovery. This Web-based software environment may be useful for the validation and analysis of functional associations in gene subsets identified by high-throughput experiments.
Figures



References
-
- Bremer E, Hakenberg J, Han EH, Berrar D, Dubitzky W, editor. Knowledge Discovery in Life Science Literature. Vol. 3886. Lecture Notes in Computer Science, Berlin: Springer; 2006. http://www.springerlink.com/content/th9635n15671
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
