

Identification of diagnostic subnetwork markers for cancer in human protein-protein interaction network
- PMID: 20946619
- PMCID: PMC3026382
- DOI: 10.1186/1471-2105-11-S6-S8
Identification of diagnostic subnetwork markers for cancer in human protein-protein interaction network
Abstract
Background: Finding reliable gene markers for accurate disease classification is very challenging due to a number of reasons, including the small sample size of typical clinical data, high noise in gene expression measurements, and the heterogeneity across patients. In fact, gene markers identified in independent studies often do not coincide with each other, suggesting that many of the predicted markers may have no biological significance and may be simply artifacts of the analyzed dataset. To find more reliable and reproducible diagnostic markers, several studies proposed to analyze the gene expression data at the level of groups of functionally related genes, such as pathways. Studies have shown that pathway markers tend to be more robust and yield more accurate classification results. One practical problem of the pathway-based approach is the limited coverage of genes by currently known pathways. As a result, potentially important genes that play critical roles in cancer development may be excluded. To overcome this problem, we propose a novel method for identifying reliable subnetwork markers in a human protein-protein interaction (PPI) network.
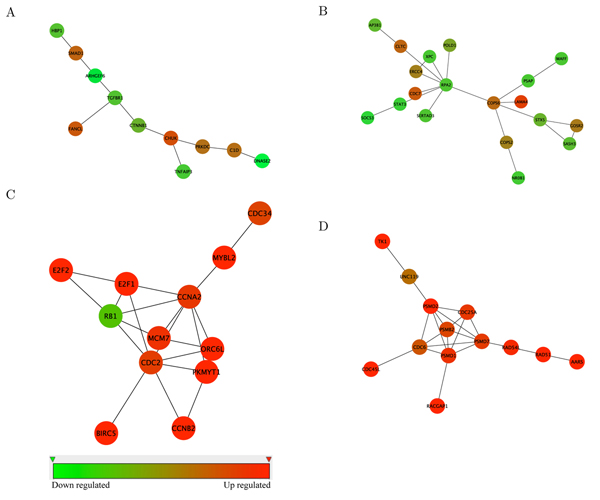
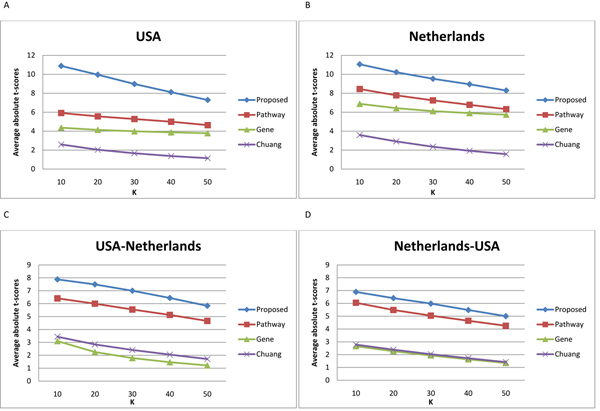
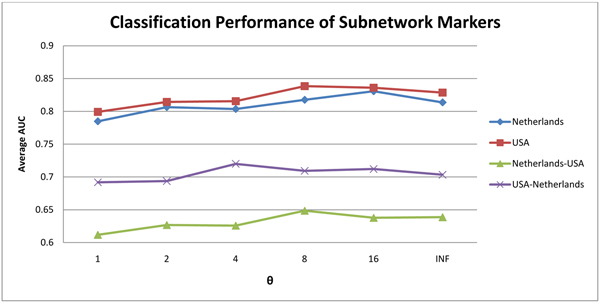
Results: In this method, we overlay the gene expression data with the PPI network and look for the most discriminative linear paths that consist of discriminative genes that are highly correlated to each other. The overlapping linear paths are then optimally combined into subnetworks that can potentially serve as effective diagnostic markers. We tested our method on two independent large-scale breast cancer datasets and compared the effectiveness and reproducibility of the identified subnetwork markers with gene-based and pathway-based markers. We also compared the proposed method with an existing subnetwork-based method.
Conclusions: The proposed method can efficiently find reliable subnetwork markers that outperform the gene-based and pathway-based markers in terms of discriminative power, reproducibility and classification performance. Subnetwork markers found by our method are highly enriched in common GO terms, and they can more accurately classify breast cancer metastasis compared to markers found by a previous method.
Figures




References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
