

Dent's disease
- PMID: 20946626
- PMCID: PMC2964617
- DOI: 10.1186/1750-1172-5-28
Dent's disease
Abstract
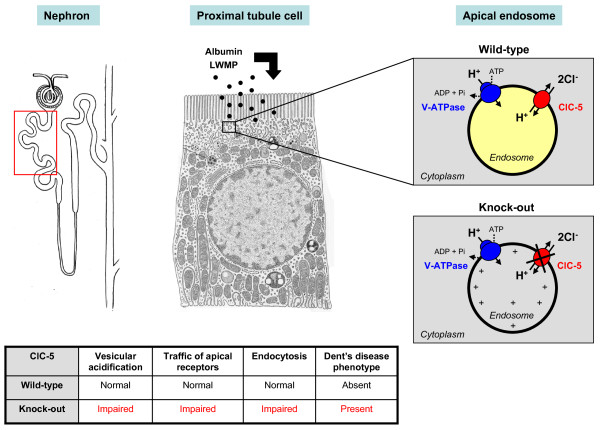
Dent's disease is a renal tubular disorder characterized by manifestations of proximal tubule dysfunction, including low-molecular-weight proteinuria, hypercalciuria, nephrolithiasis, nephrocalcinosis, and progressive renal failure. These features are generally found in males only, and may be present in early childhood, whereas female carriers may show a milder phenotype. Prevalence is unknown; the disorder has been reported in around 250 families to date. Complications such as rickets or osteomalacia may occur. The disease is caused by mutations in either the CLCN5 (Dent disease 1) or OCRL1 (Dent disease 2) genes that are located on chromosome Xp11.22 and Xq25, respectively. CLCN5 encodes the electrogenic Cl⁻/H(+) exchanger ClC-5, which belongs to the CLC family of Cl⁻ channels/transporters. OCRL1 encodes a phosphatidylinositol bisphosphate (PIP₂) 5-phosphatase and mutations are also associated with Lowe Syndrome. The phenotype of Dent's disease is explained by the predominant expression of ClC-5 in the proximal tubule segments of the kidney. No genotype-phenotype correlation has been described thus far, and there is considerable intra-familial variability in disease severity. A few patients with Dent's disease do not harbour mutations in CLCN5 and OCRL1, pointing to the involvement of other genes. Diagnosis is based on the presence of all three of the following criteria: low-molecular-weight proteinuria, hypercalciuria and at least one of the following: nephrocalcinosis, kidney stones, hematuria, hypophosphatemia or renal insufficiency. Molecular genetic testing confirms the diagnosis. The differential diagnosis includes other causes of generalized dysfunction of the proximal tubules (renal Fanconi syndrome), hereditary, acquired, or caused by exogenous substances. Antenatal diagnosis and pre-implantation genetic testing is not advised. The care of patients with Dent's disease is supportive, focusing on the treatment of hypercalciuria and the prevention of nephrolithiasis. The vital prognosis is good in the majority of patients. Progression to end-stage renal failure occurs between the 3rd and 5th decades of life in 30-80% of affected males.
Figures
References
-
- Wrong OM, Norden AGW, Feest TG. Dent's disease; a familial proximal renal tubular syndrome with low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease, progressive renal failure and a marked male predominance. Q J Med. 1994;87:473–493. - PubMed
-
- Lloyd SE, Pearce SH, Fisher SE, Steinmeyer K, Schwappach B, Scheinman SJ, Harding B, Bolino A, Devoto M, Goodyer P, Rigden SP, Wrong O, Jentsch TJ, Craig IW, Thakker RV. A common molecular basis for three inherited kidney stone diseases. Nature. 1996;379:445–449. doi: 10.1038/379445a0. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
