

Review
doi: 10.1007/s00467-010-1637-4.
Epub 2010 Oct 15.
Advances in our understanding of the pathogenesis of glomerular thrombotic microangiopathy
Affiliations
- PMID: 20949284
- PMCID: PMC3043262
- DOI: 10.1007/s00467-010-1637-4
Item in Clipboard
Review
Advances in our understanding of the pathogenesis of glomerular thrombotic microangiopathy
Pediatr Nephrol.
2011 Apr.
Abstract
Glomerular thrombotic microangiopathy is a hallmark feature of haemolytic uraemic syndrome, the leading cause of acute renal failure in childhood. This paper is a review of the different mechanistic pathways that lead to this histological picture in the kidney. It will focus on atypical HUS and complement dysregulation, but will also highlight some other recent advances in our understanding of this condition, including the potential role of the molecule vascular endothelial growth factor-A (VEGF-A).
Figures
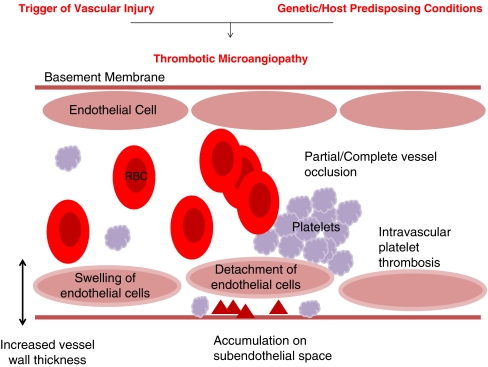
Thrombotic microangiopathy: “the final events”. Thrombotic microangiopathy is the pathological process that is the final common pathway of many disease processes, but is most commonly associated with haemolytic uraemic syndrom (HUS) and thrombotic thrombocytopaenic purpura (TTP). Pathological features include: increased vessel wall thickness, swelling and/or detachment of endothelial cells, platelet aggregation/thrombosis, accumulation of debris/material in subendothelial space and partial or complete vessel occlusion. Clinically: microangiopathic haemolytic anaemia—sheer stress from vessel lumen occlusion produces fragmented red cells, thrombocytopaenia—platelets are consumed by local thrombosis and organ ischaemia—renal failure or neurological symptoms
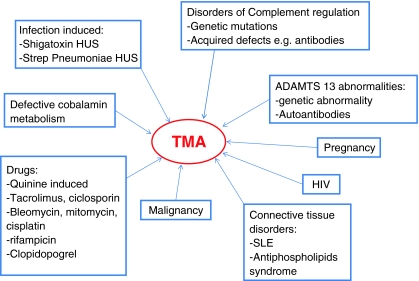
Precipitants of thrombotic microangiopathy (TMA). SLE Systemic lupus erythematosus
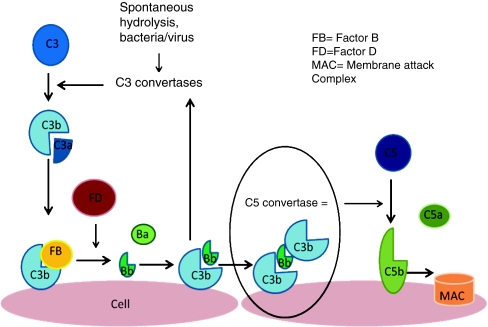
The alternative pathway is triggered by the covalent binding of C3b to a pathogen or cell surface. Next, factor B binds to surface bond C3b, making it susceptible to plasma factor D cleavage. The result is production of Ba and active protease Bb, which remains bound to C3b creating C3bBb, which is the C3 convertase of the alternative complement pathway. This starts the amplification loop with C3 convertase generating more C3b on the cell surface and the process repeats. Ultimately, there will be C3b saturation on the cell surface with release of C3a, a small inflammatory mediator. Eventually, some of the C3b binds to pre-existing C3 convertase producing C3b2Bb, which is the alternative pathway’s C5 convertase. This cleaves C5 into C5b, which generates the membrane attack complex (MAC), and C5a, a potent pro-inflammatory mediator. Complement-mediated endothelial cell injury creates a prothrombotic state. It exposes subendothelial collagens and releases von Willebrand factor and fibrinogen formation
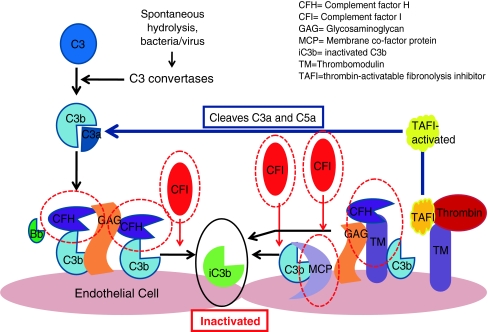
Complement regulators are shown circled in red. These include factors H and I and membrane co-factor protein. Each acts to promote the inactivation of C3b and prevent further progression of the complement cascade. Factor H binds Cb3 and works with factor I to inactivate it. Both complement factors H and I are serum-based. Membrane co-factor protein is cell-bound. It also binds to C3b, which has become attached to cells and works with factor I to inactivate it. Thrombomodulin is also shown as mutations and has been associated with atypical HUS. It regulates complement by acting to inactivate the proinflammatory mediators C3a and C5a and accelerating factor I-mediated C3b inactivation. It also plays a role in local coagulation regulation through its interactions with thrombin
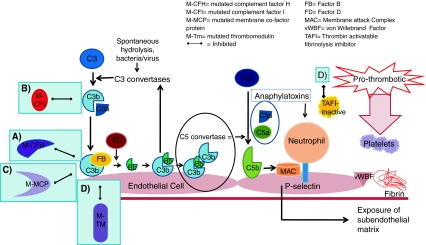
Development of thrombotic microangiopathy with loss of normal complement regulatory factor function. Loss of alternative complement pathway regulation caused by loss of function of factors H and I, membrane co-factor protein or thrombomodulin results in complement activation directed against endothelial cells. a Mutated complement factor H cannot bind to cell surface C3b to stop alternative complement pathway attack. b Mutated complement factor I cannot cleave C3b. c Mutated membrane co-factor protein cannot work with factor I to degrade C3b. d Mutated thrombomodulin cannot bind C3b and accelerate factor I-medicated inactivation of C3b. It cannot activate thrombin activatable fibrinolysis inhibitor (TAFI), which inactivates anaphylatoxins C3a and C5a. It also cannot bind thrombin. With the loss of these regulatory functions of each of these factors C3b is not inactivated and progression of the complement cascade ensues. C3b then binds with Bb and then further C3b bind to this complex, which forms C5 convertase. This cleaves C5 to C5b and the resulting cascade results in formation of the membrane attack complex (MAC). C3a and C5a act as anaphylatoxins, which attract neutrophils to the cells under complement attack. Activating mutations of factor B and C3b also result in progression of the complement pathway. The result of any of these mutations is endothelial cell damage, causing exposure of the subendothelial matrix. This results in a prothrombotic state with fibrin deposition and release of von Willebrand factor, which attracts platelets to the site. Platelets will aggregate. The result is a full complement attack and thrombus formation. Partial or complete vessel occlusion will result. This will produce platelet consumption and red cell damage and TMA is the result
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
