

De novo mutations in FOXP1 in cases with intellectual disability, autism, and language impairment
- PMID: 20950788
- PMCID: PMC2978954
- DOI: 10.1016/j.ajhg.2010.09.017
De novo mutations in FOXP1 in cases with intellectual disability, autism, and language impairment
Abstract
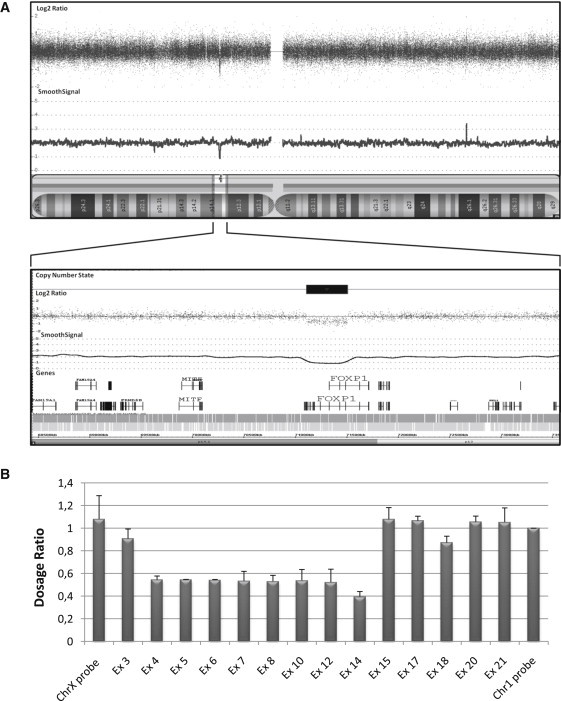
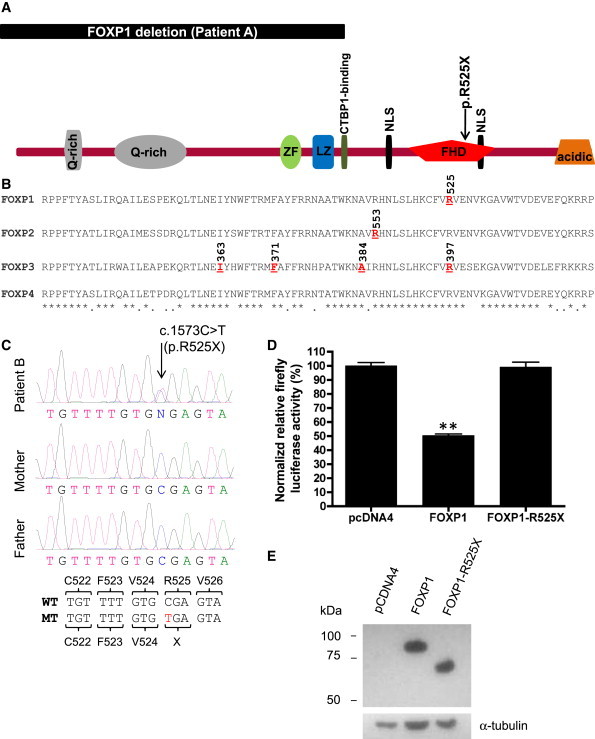
Heterozygous mutations in FOXP2, which encodes a forkhead transcription factor, have been shown to cause developmental verbal dyspraxia and language impairment. FOXP2 and its closest homolog, FOXP1, are coexpressed in brain regions that are important for language and cooperatively regulate developmental processes, raising the possibility that FOXP1 may also be involved in developmental conditions that are associated with language impairment. In order to explore this possibility, we searched for mutations in FOXP1 in patients with intellectual disability (ID; mental retardation) and/or autism spectrum disorders (ASD). We first performed array-based genomic hybridization on sporadic nonsyndromic ID (NSID) (n = 30) or ASD (n = 80) cases. We identified a de novo intragenic deletion encompassing exons 4-14 of FOXP1 in a patient with NSID and autistic features. In addition, sequencing of all coding exons of FOXP1 in sporadic NSID (n = 110) or ASD (n = 135) cases, as well as in 570 controls, revealed the presence of a de novo nonsense mutation (c.1573C>T [p.R525X]) in the conserved forkhead DNA-binding domain in a patient with NSID and autism. Luciferase reporter assays showed that the p.R525X alteration disrupts the activity of the protein. Formal assessments revealed that both patients with de novo mutations in FOXP1 also show severe language impairment, mood lability with physical aggressiveness, and specific obsessions and compulsions. In conclusion, both FOXP1 and FOXP2 are associated with language impairment, but decrease of the former has a more global impact on brain development than that of the latter.
Copyright © 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Fisher S.E., Lai C.S., Monaco A.P. Deciphering the genetic basis of speech and language disorders. Annu. Rev. Neurosci. 2003;26:57–80. - PubMed
-
- Fisher S.E., Scharff C. FOXP2 as a molecular window into speech and language. Trends Genet. 2009;25:166–177. - PubMed
-
- Enard W., Gehre S., Hammerschmidt K., Hölter S.M., Blass T., Somel M., Brückner M.K., Schreiweis C., Winter C., Sohr R. A humanized version of Foxp2 affects cortico-basal ganglia circuits in mice. Cell. 2009;137:961–971. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
