

Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase
- PMID: 20951943
- PMCID: PMC2957477
- DOI: 10.1016/j.ccr.2010.08.012
Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase
Abstract
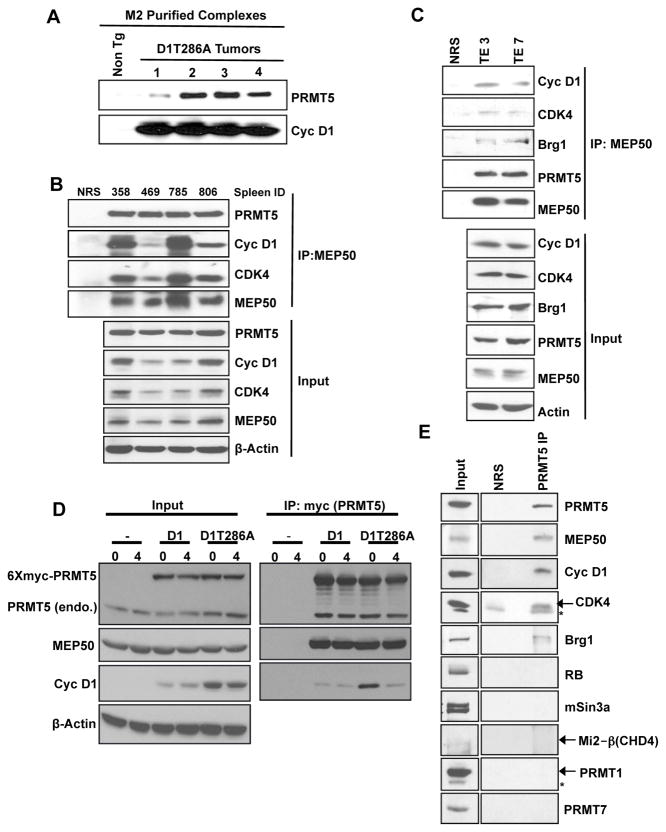
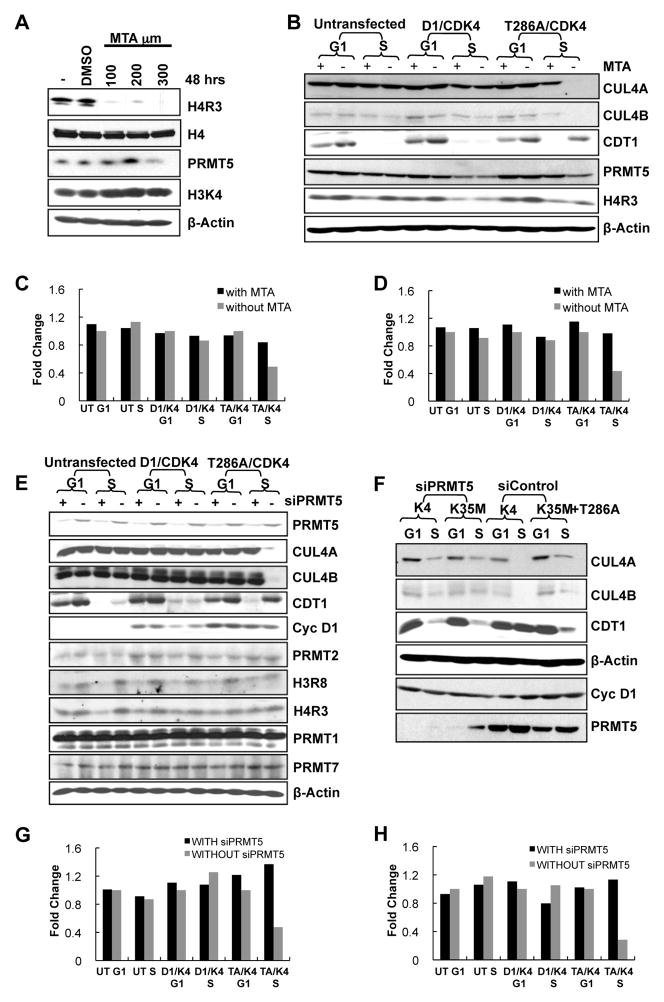
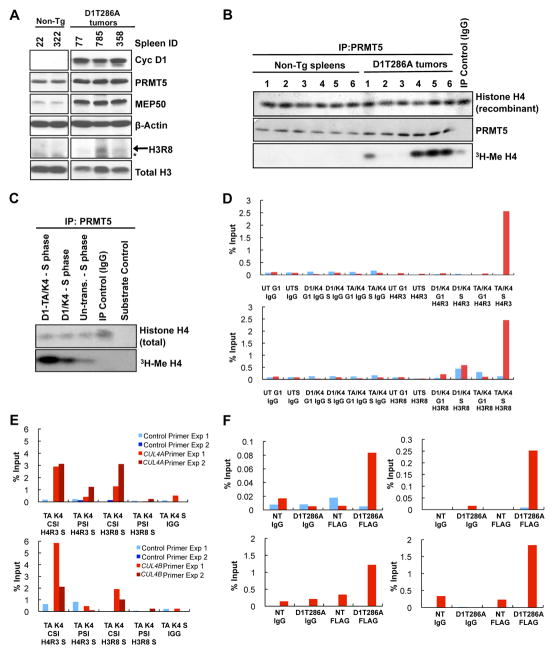
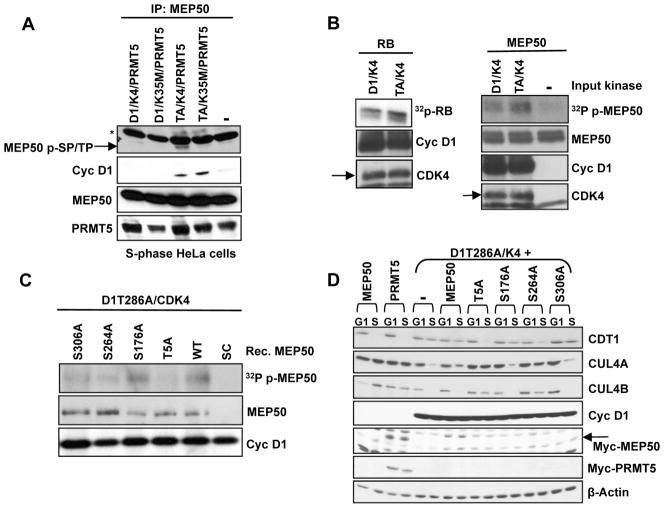
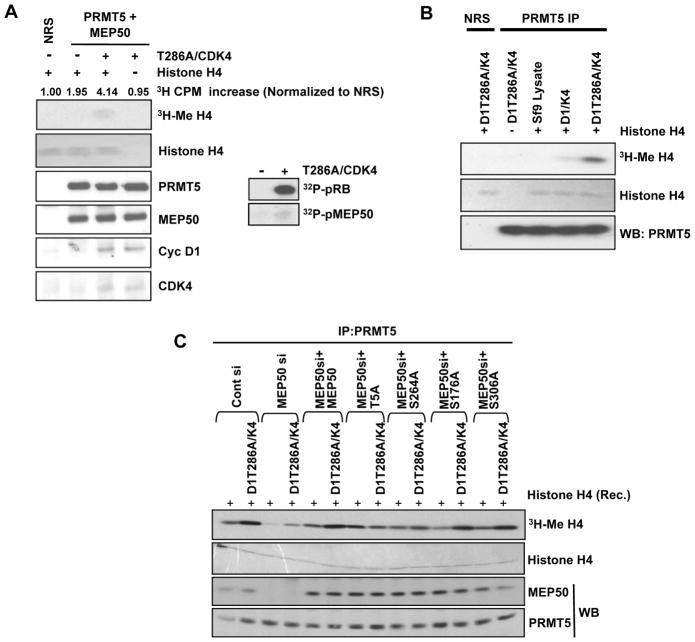
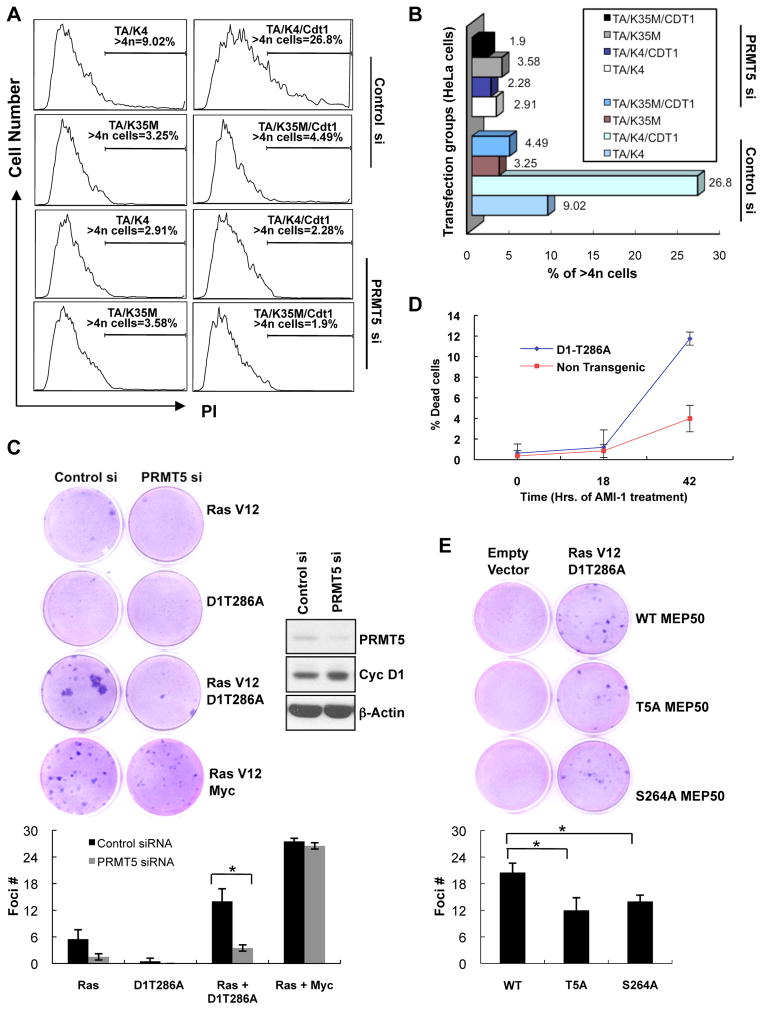
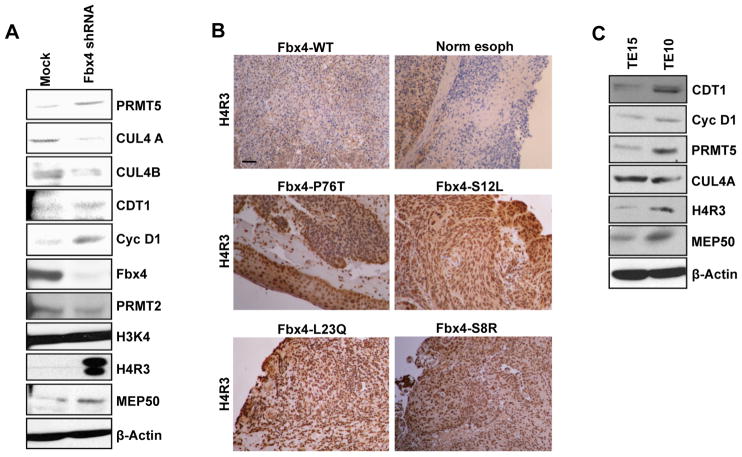
Cyclin D1 elicits transcriptional effects through inactivation of the retinoblastoma protein and direct association with transcriptional regulators. The current work reveals a molecular relationship between cyclin D1/CDK4 kinase and protein arginine methyltransferase 5 (PRMT5), an enzyme associated with histone methylation and transcriptional repression. Primary tumors of a mouse lymphoma model exhibit increased PRMT5 methyltransferase activity and histone arginine methylation. Analyses demonstrate that MEP50, a PRMT5 coregulatory factor, is a CDK4 substrate, and phosphorylation increases PRMT5/MEP50 activity. Increased PRMT5 activity mediates key events associated with cyclin D1-dependent neoplastic growth, including CUL4 repression, CDT1 overexpression, and DNA rereplication. Importantly, human cancers harboring mutations in Fbx4, the cyclin D1 E3 ligase, exhibit nuclear cyclin D1 accumulation and increased PRMT5 activity.
Copyright © 2010 Elsevier Inc. All rights reserved.
Figures
References
-
- Aggarwal P, Lessie MD, Lin DI, Pontano L, Gladden AB, Nuskey B, Goradia A, Wasik MA, Klein-Szanto AJ, Rustgi AK, et al. Nuclear accumulation of cyclin D1 during S phase inhibits Cul4-dependent Cdt1 proteolysis and triggers p53-dependent DNA rereplication. Genes Dev. 2007;21:2908–2922. - PMC - PubMed
-
- Bani-Hani K, Martin IG, Hardie LJ, Mapstone N, Briggs JA, Forman D, Wild CP. Prospective study of cyclin D1 overexpression in Barrett’s esophagus: association with increased risk of adenocarcinoma. J Natl Cancer Inst. 2000;92:1316–1321. - PubMed
-
- Bartkova J, Lukas J, Muller H, Lutzhoft D, Strauss M, Bartek J. Cyclin D1 protein expression and function in human breast cancer. Int J Cancer. 1994a;57:353–361. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
