

Functional proteomic analysis for regulatory T cell surveillance of the HIV-1-infected macrophage
- PMID: 20954747
- PMCID: PMC3108468
- DOI: 10.1021/pr1009178
Functional proteomic analysis for regulatory T cell surveillance of the HIV-1-infected macrophage
Abstract
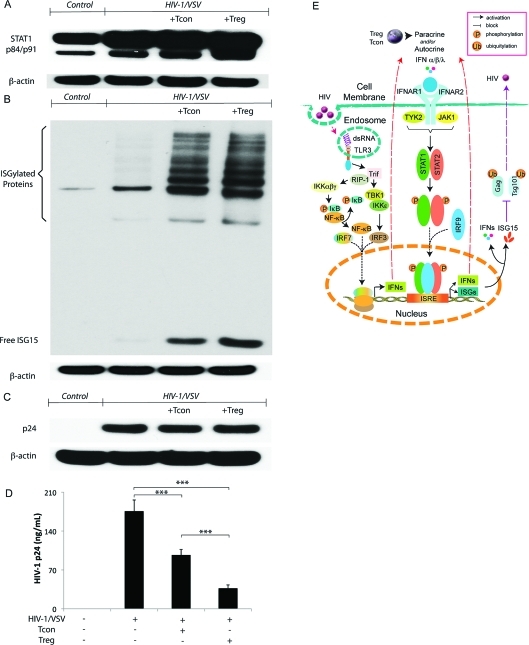
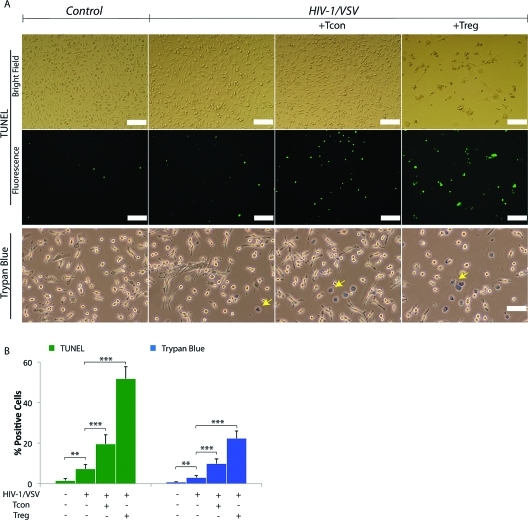
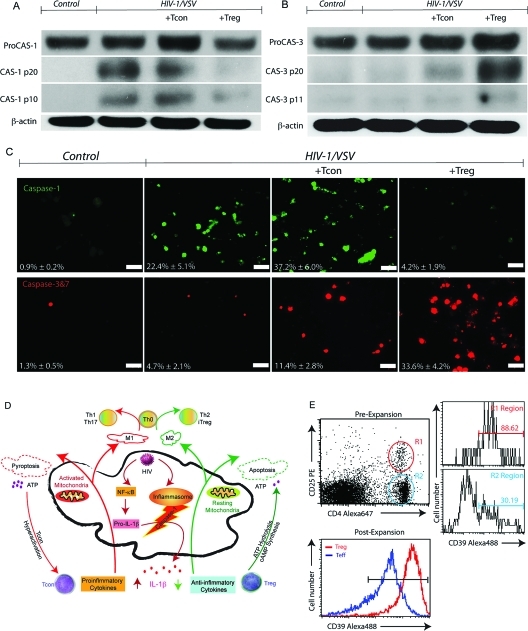
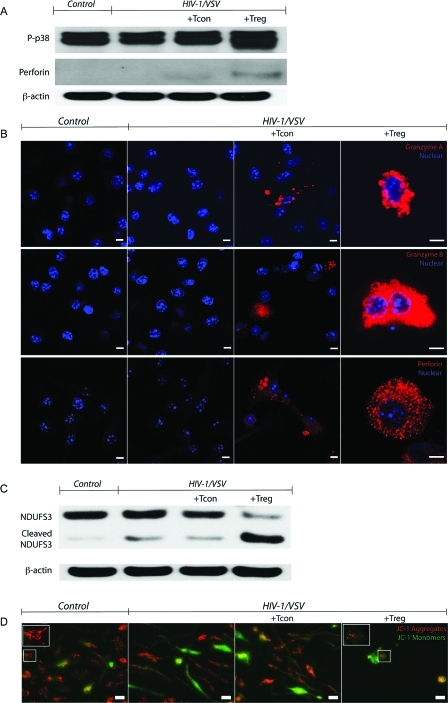
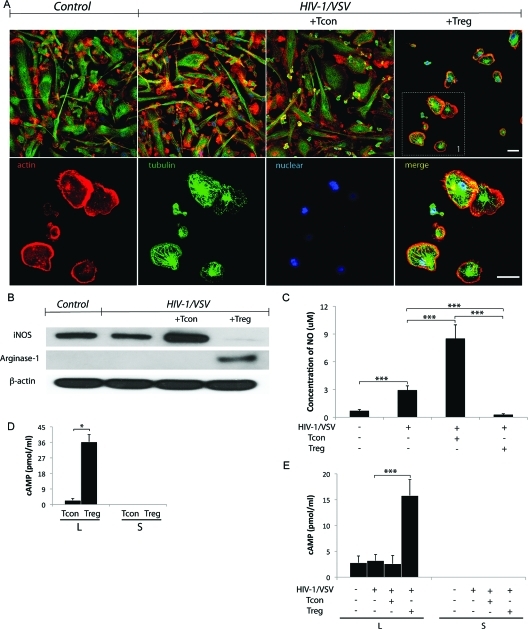
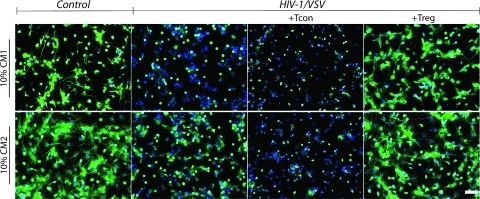
Regulatory T cells (Treg) induce robust neuroprotection in murine models of neuroAIDS, in part, through eliciting anti-inflammatory responses for HIV-1-infected brain mononuclear phagocytes (MP; macrophage and microglia). Herein, using both murine and human primary cell cultures in proteomic and cell biologic tests, we report that Treg promotes such neuroprotection by an even broader range of mechanisms than previously seen including inhibition of virus release, killing infected MP, and inducing phenotypic cell switches. Changes in individual Treg-induced macrophage proteins were quantified by iTRAQ labeling followed by mass spectrometry identifications. Reduction in virus release paralleled the upregulation of interferon-stimulated gene 15, an ubiquitin-like protein involved in interferon-mediated antiviral immunity. Treg killed virus-infected macrophages through caspase-3 and granzyme and perforin pathways. Independently, Treg transformed virus-infected macrophages from an M1 to an M2 phenotype by down- and up- regulation of inducible nitric oxide synthase and arginase 1, respectively. Taken together, Treg affects a range of virus-infected MP functions. The observations made serve to challenge the dogma of solitary Treg immune suppressor functions and provides novel insights into how Treg affects adaptive immunosurveillance for control of end organ diseases, notably neurocognitive disorders associated with advanced viral infection.
Figures
References
-
- Robertson K. R.; Smurzynski M.; Parsons T. D.; Wu K.; Bosch R. J.; Wu J.; McArthur J. C.; Collier A. C.; Evans S. R.; Ellis R. J. The prevalence and incidence of neurocognitive impairment in the HAART era. AIDS 2007, 21 (14), 1915–21. - PubMed
-
- Antinori A.; Arendt G.; Becker J. T.; Brew B. J.; Byrd D. A.; Cherner M.; Clifford D. B.; Cinque P.; Epstein L. G.; Goodkin K.; Gisslen M.; Grant I.; Heaton R. K.; Joseph J.; Marder K.; Marra C. M.; McArthur J. C.; Nunn M.; Price R. W.; Pulliam L.; Robertson K. R.; Sacktor N.; Valcour V.; Wojna V. E. Updated research nosology for HIV-associated neurocognitive disorders. Neurology 2007, 69 (18), 1789–99. - PMC - PubMed
-
- McArthur J. C.; Brew B. J.; Nath A. Neurological complications of HIV infection. Lancet Neurol. 2005, 4 (9), 543–55. - PubMed
-
- Everall I. P.; Hansen L. A.; Masliah E. The shifting patterns of HIV encephalitis neuropathology. Neurotox. Res. 2005, 8 (1−2), 51–61. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
