Figure 1
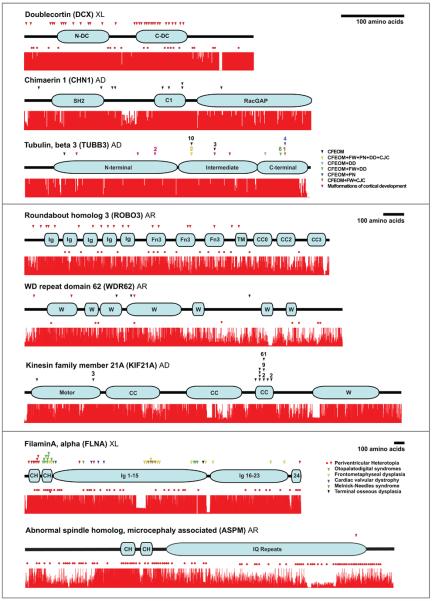
Schematics of each protein are illustrated with the name of the protein and its abbreviation to the upper left followed by mode of inheritance of disease (XL = X-linked, AD = autosomal dominant, AR = autosomal recessive). Proteins are ordered according to length with the shortest on the top and the longest on the bottom, and are presented in three differently scaled groups, each based on the amino acid scale bar found to the upper right of the group. ‘Stop’ mutations (including nonsense, frameshift, intragenic deletions, or splicing alleles that cause frameshifts) are represented by red dots positioned below the protein corresponding to the location of the mutation. Missense mutations are represented by arrowheads positioned above the protein corresponding to the location of the mutation. Each dot and arrowhead represents a unique mutation. Additional arrowheads aligned directly above (or red dots aligned directly below) one another indicate distinct mutations have been reported at the same amino acid residue (i.e. in KIF21A, the mutation altering an amino acid residue in the distal motor domain has been identified in three unrelated patients). Some arrowheads or red dots are unevenly stacked; they represent unique mutations that, for reasons of scale, were not able to be aligned horizontally along the protein. When multiple unrelated patients have been reported with the identical mutation, the number of times that mutation has been independently observed appears above the arrow (or below the red dot). Within each protein, each arrowhead color corresponds to a distinct disease phenotype, and red arrowheads mean that the missense mutation results in the same phenotype as the Stop mutation(s). When more than one phenotype results from mutations in a given protein, the arrowheads/circles are color-coded with a corresponding key to the right of the protein. For example, in the intermediate domain of TUBB3, 10 unrelated patients with the same phenotype have been reported to harbor the identical missense mutation, while 2 other unrelated patients share a distinct phenotype and a different missense mutation that alters the same amino acid but to a different residue. Conservation tracts were made by aligning the most homologous amino acid sequences available for each gene for human, chimpanzee, dog, mouse, chicken, and zebrafish using the ClustalX software package with default settings. Accession numbers of the sequences used are listed below. Complete sequence data for FLNA, KIF21A and ROBO3 was not available for chimpanzee and so rhesus monkey FLNA sequence, and orangutan KIF21a and ROBO3 sequences were substituted. Chicken ROBO3 and WDR62 sequences as well as zebrafish DCX sequence were not available and no substitutions for these were made. Several sequences for CHN1 and ROBO3 were predicted using the UCSC genome browser multiple sequence alignment tool based on the comparative genomics conservation tracts (uc# noted below) and the dog sequence for KIF21a was predicted from genome sequence with N-SCAN. Protein motifs, key abbreviations and sequence accession numbers for conservation tracts Doublecortin (DCX) Protein motifs: N-terminal and C-terminal doublecortin domains; Accession numbers: NP_000546.2, XP_529107.2, XP_853182.1, NP_034155.2, NP_989666.1. Chimaerin 1 (CHN1) Protein motifs: N-terminal Src homology 2 (SH2), phorbol esters/diacylglycerol binding (C1), and Rac GTPase activating (RacGAP) domains; Accession numbers: uc002uji.1_hg18, uc002uji.1_panTro2, uc002uji.1_canFam2, uc002uji.1_mm9, NP_001012970.1, NP_998165.1. Tubulin, beta 3 (TUBB3) Key abbreviations: CFEOM (congenital fibrosis of the extraocular muscles), FW (facial weakness), PN (peripheral neuropathy), DD (intellectual and social developmental delay), and CJC (congenital joint contractures); Accession numbers: NP_006077.2, NP_001038974.1, XP_860049.1, NP_075768.1, NP_998655.1, NP_001026769.1. Roundabout homolog 3 (ROBO3) Protein motifs: Immunoglobulin-like (Ig), fibronectin-like (Fn3), transmembrane, and conserved cytoplasmic (CC) domains; Accession numbers: ROBO3 NP_071765.2, uc001qbc.1_ponAbe2, XP_546425.2, XP_001476890.1, NP_571557.1. WD repeat domain 62 (WDR62) Protein motifs: WD40 (W) domains; Accession numbers: NP_001077430.1, XP_512609.2, XP_853669.1, XP_896469.1, XP_699579.2. Kinesin family member 21A (KIF21A) Protein motifs: Motor (M), coiled-coil (CC), and WD40 (W) domains; Accession numbers: NP_060111.2, XP_002823145.1, N-SCAN (chr27.18.003.a), NP_060111.2, XP_001920258.1, XP_415936.2. FilaminA, alpha (FLNA) Protein motifs: Calpain-homology (CH) and immunoglobulin-like (Ig) domains; Accession numbers: NP_001104026.1, XP_001091073.1, XP_867483.1, NP_034357.2, NP_989905.1, XP_001922206.1. Abnormal spindle homolog, microcephaly associated (ASPM) Protein motifs: Calpainhomology (CH) and isoleucine-glutamine repeat (IQ) domains; Accession numbers: NP_060606.3, NP_001008994.1, XP_537130.2, NP_033921.3, XP_001923712.1, XP_422197.2.