

Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema
- PMID: 20956295
- PMCID: PMC2973911
- DOI: 10.1073/pnas.1005574107
Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema
Abstract
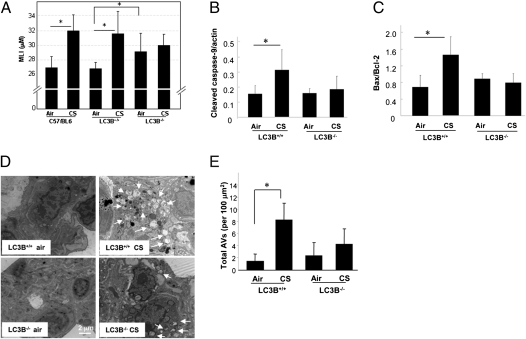
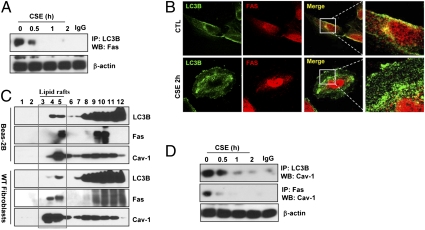
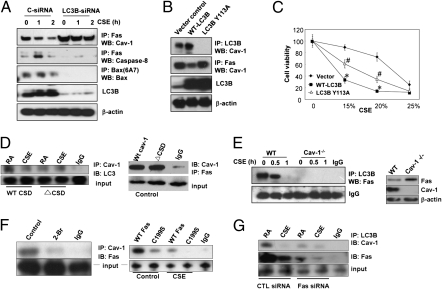
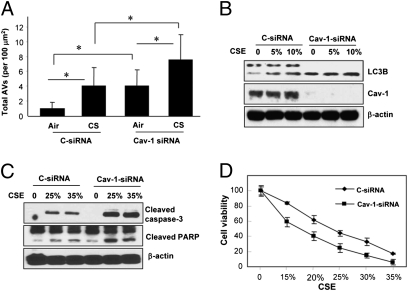
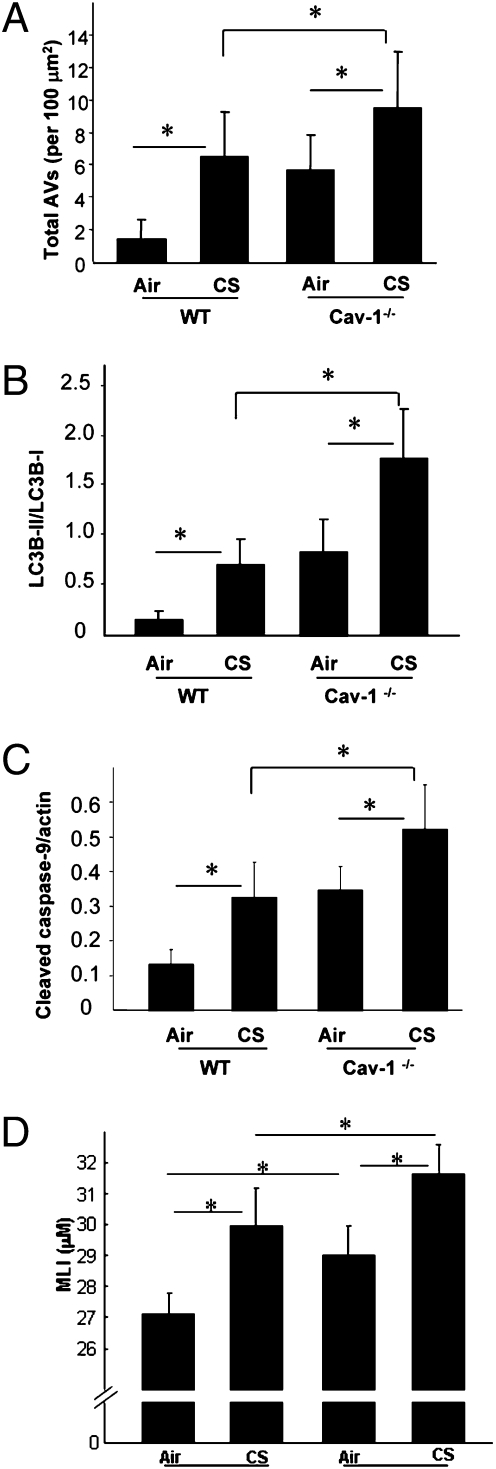
Chronic obstructive pulmonary disease (COPD) is a debilitating disease caused by chronic exposure to cigarette smoke (CS), which involves airway obstruction and alveolar loss (i.e., emphysema). The mechanisms of COPD pathogenesis remain unclear. Our previous studies demonstrated elevated autophagy in human COPD lung, and as a cellular and tissue response to CS exposure in an experimental model of emphysema in vivo. We identified the autophagic protein microtubule-associated protein 1 light chain-3B (LC3B) as a positive regulator of CS-induced lung epithelial cell death. We now extend these initial observations to explore the mechanism by which LC3B mediates CS-induced apoptosis and emphysema development in vivo. Here, we observed that LC3B(-/-) mice had significantly decreased levels of apoptosis in the lungs after CS exposure, and displayed resistance to CS-induced airspace enlargement, relative to WT littermate mice. We found that LC3B associated with the extrinsic apoptotic factor Fas in lipid rafts in an interaction mediated by caveolin-1 (Cav-1). The siRNA-dependent knockdown of Cav-1 sensitized epithelial cells to CS-induced apoptosis, as evidenced by enhanced death-inducing signaling complex formation and caspase activation. Furthermore, Cav-1(-/-) mice exhibited higher levels of autophagy and apoptosis in the lung in response to chronic CS exposure in vivo. In conclusion, we demonstrate a pivotal role for the autophagic protein LC3B in CS-induced apoptosis and emphysema, suggestive of novel therapeutic targets for COPD treatment. This study also introduces a mechanism by which LC3B, through interactions with Cav-1 and Fas, can regulate apoptosis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Lopez AD, Murray CC. The global burden of disease, 1990-2020. Nat Med. 1998;4:1241–1243. - PubMed
-
- Rabe KF, et al. Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;176:532–555. - PubMed
-
- Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev. 2007;87:1047–1082. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
