

Requirement of the ATM/p53 tumor suppressor pathway for glucose homeostasis
- PMID: 20956556
- PMCID: PMC3004274
- DOI: 10.1128/MCB.00347-10
Requirement of the ATM/p53 tumor suppressor pathway for glucose homeostasis
Abstract
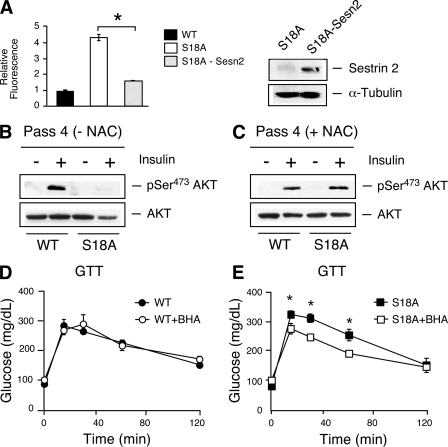
Ataxia telangiectasia (A-T) patients can develop multiple clinical pathologies, including neuronal degeneration, an elevated risk of cancer, telangiectasias, and growth retardation. Patients with A-T can also exhibit an increased risk of insulin resistance and type 2 diabetes. The ATM protein kinase, the product of the gene mutated in A-T patients (Atm), has been implicated in metabolic disease, which is characterized by insulin resistance and increased cholesterol and lipid levels, blood pressure, and atherosclerosis. ATM phosphorylates the p53 tumor suppressor on a site (Ser15) that regulates transcription activity. To test whether the ATM pathway that regulates insulin resistance is mediated by p53 phosphorylation, we examined insulin sensitivity in mice with a germ line mutation that replaces the p53 phosphorylation site with alanine. The loss of p53 Ser18 (murine Ser15) led to increased metabolic stress, including severe defects in glucose homeostasis. The mice developed glucose intolerance and insulin resistance. The insulin resistance correlated with the loss of antioxidant gene expression and decreased insulin signaling. N-Acetyl cysteine (NAC) treatment restored insulin signaling in late-passage primary fibroblasts. The addition of an antioxidant in the diet rendered the p53 Ser18-deficient mice glucose tolerant. This analysis demonstrates that p53 phosphorylation on an ATM site is an important mechanism in the physiological regulation of glucose homeostasis.
Figures
References
-
- Armata, H. L., D. S. Garlick, and H. K. Sluss. 2007. The ataxia telangiectasia-mutated target site Ser18 is required for p53-mediated tumor suppression. Cancer Res. 67:11696-11703. - PubMed
-
- Balaban, R. S., S. Nemoto, and T. Finkel. 2005. Mitochondria, oxidants, and aging. Cell 120:483-495. - PubMed
-
- Bar, R. S., W. R. Levis, M. M. Rechler, L. C. Harrison, C. Siebert, J. Podskalny, J. Roth, and M. Muggeo. 1978. Extreme insulin resistance in ataxia telangiectasia: defect in affinity of insulin receptors. N. Engl. J. Med. 298:1164-1171. - PubMed
-
- Barlow, C., S. Hirotsune, R. Paylor, M. Liyanage, M. Eckhaus, F. Collins, Y. Shiloh, J. N. Crawley, T. Ried, D. Tagle, and A. Wynshaw-Boris. 1996. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86:159-171. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous