

Mitochondria and neuroplasticity
- PMID: 20957078
- PMCID: PMC2949087
- DOI: 10.1042/AN20100019
Mitochondria and neuroplasticity
Abstract
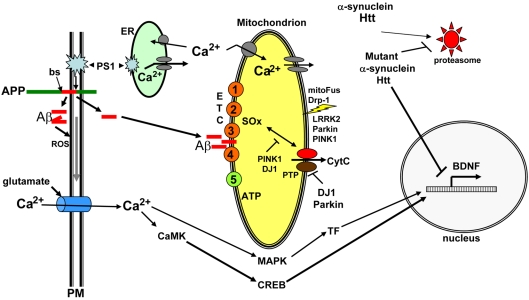
The production of neurons from neural progenitor cells, the growth of axons and dendrites and the formation and reorganization of synapses are examples of neuroplasticity. These processes are regulated by cell-autonomous and intercellular (paracrine and endocrine) programs that mediate responses of neural cells to environmental input. Mitochondria are highly mobile and move within and between subcellular compartments involved in neuroplasticity (synaptic terminals, dendrites, cell body and the axon). By generating energy (ATP and NAD(+)), and regulating subcellular Ca(2+) and redox homoeostasis, mitochondria may play important roles in controlling fundamental processes in neuroplasticity, including neural differentiation, neurite outgrowth, neurotransmitter release and dendritic remodelling. Particularly intriguing is emerging data suggesting that mitochondria emit molecular signals (e.g. reactive oxygen species, proteins and lipid mediators) that can act locally or travel to distant targets including the nucleus. Disturbances in mitochondrial functions and signalling may play roles in impaired neuroplasticity and neuronal degeneration in Alzheimer's disease, Parkinson's disease, psychiatric disorders and stroke.
Keywords: AD, Alzheimer's disease; AP, adaptor protein; APP, amyloid precursor protein; Aβ, amyloid β-peptide; BDNF, brain-derived neurotrophic factor; CR, caloric restriction; CREB, cAMP-response-element-binding protein; CaMK, Ca2+/calmodulin-dependent protein kinase; ES, embryonic stem; ETC, electron transport chain; HD, Huntington's disease; LRRK2, leucine-rich repeat kinase 2; LTP, long-term potentiation; MAPK, mitogen-activated protein kinase; Mn-SOD, manganese superoxide dismutase; NGF, nerve growth factor; NMDA, N-methyl-d-aspartate; Nrf1, nuclear respiratory factor 1; OPA1, Optic Atrophy-1; PD, Parkinson's disease; PGC1α, peroxisome-proliferator-activated receptor γ co-activator 1α; PINK1, PTEN (phosphatase and tensin homologue deleted on chromosome 10)-induced kinase 1; PPAR, peroxisome-proliferator-activated receptor; UCP, uncoupling protein; mitochondria biogenesis; mitochondria fission and fusion; neural progenitor cell.
Figures
Similar articles
-
Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease.Free Radic Biol Med. 2013 Sep;62:90-101. doi: 10.1016/j.freeradbiomed.2012.11.014. Epub 2012 Nov 29. Free Radic Biol Med. 2013. PMID: 23200807 Free PMC article.
-
Mitochondrial diseases of the brain.Free Radic Biol Med. 2013 Oct;63:1-29. doi: 10.1016/j.freeradbiomed.2013.03.018. Epub 2013 Apr 6. Free Radic Biol Med. 2013. PMID: 23567191 Review.
-
Impaired autophagy and APP processing in Alzheimer's disease: The potential role of Beclin 1 interactome.Prog Neurobiol. 2013 Jul-Aug;106-107:33-54. doi: 10.1016/j.pneurobio.2013.06.002. Epub 2013 Jul 1. Prog Neurobiol. 2013. PMID: 23827971 Review.
-
Adaptive responses of neuronal mitochondria to bioenergetic challenges: Roles in neuroplasticity and disease resistance.Free Radic Biol Med. 2017 Jan;102:203-216. doi: 10.1016/j.freeradbiomed.2016.11.045. Epub 2016 Nov 29. Free Radic Biol Med. 2017. PMID: 27908782 Free PMC article. Review.
-
BDNF-induced local protein synthesis and synaptic plasticity.Neuropharmacology. 2014 Jan;76 Pt C:639-56. doi: 10.1016/j.neuropharm.2013.04.005. Epub 2013 Apr 16. Neuropharmacology. 2014. PMID: 23602987 Review.
Cited by
-
A new approach for the treatment of subthreshold bipolar disorders: Targeted high dose levothyroxine and repetitive transcranial magnetic stimulation for mitochondrial treatment.Front Psychiatry. 2022 Oct 13;13:976544. doi: 10.3389/fpsyt.2022.976544. eCollection 2022. Front Psychiatry. 2022. PMID: 36311500 Free PMC article. Review.
-
Neuroprotective Effect of miR-483-5p Against Cardiac Arrest-Induced Mitochondrial Dysfunction Mediated Through the TNFSF8/AMPK/JNK Signaling Pathway.Cell Mol Neurobiol. 2023 Jul;43(5):2179-2202. doi: 10.1007/s10571-022-01296-3. Epub 2022 Oct 20. Cell Mol Neurobiol. 2023. PMID: 36266523 Free PMC article.
-
Compartmentalized Signaling in Aging and Neurodegeneration.Cells. 2021 Feb 22;10(2):464. doi: 10.3390/cells10020464. Cells. 2021. PMID: 33671541 Free PMC article. Review.
-
Melatonin protects against focal cerebral ischemia-reperfusion injury in diabetic mice by ameliorating mitochondrial impairments: involvement of the Akt-SIRT3-SOD2 signaling pathway.Aging (Albany NY). 2021 Jun 11;13(12):16105-16123. doi: 10.18632/aging.203137. Epub 2021 Jun 11. Aging (Albany NY). 2021. PMID: 34118791 Free PMC article.
-
PINK1 alleviates thermal hypersensitivity in a paclitaxel-induced Drosophila model of peripheral neuropathy.PLoS One. 2020 Sep 17;15(9):e0239126. doi: 10.1371/journal.pone.0239126. eCollection 2020. PLoS One. 2020. PMID: 32941465 Free PMC article.
References
-
- Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP, Holloszy JO. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. FASEB J. 2002;16:1879–1886. - PubMed
-
- Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60:430–440. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous