

Chikungunya virus induces IPS-1-dependent innate immune activation and protein kinase R-independent translational shutoff
- PMID: 20962078
- PMCID: PMC3014158
- DOI: 10.1128/JVI.00767-10
Chikungunya virus induces IPS-1-dependent innate immune activation and protein kinase R-independent translational shutoff
Abstract
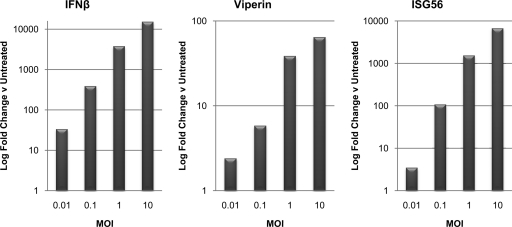
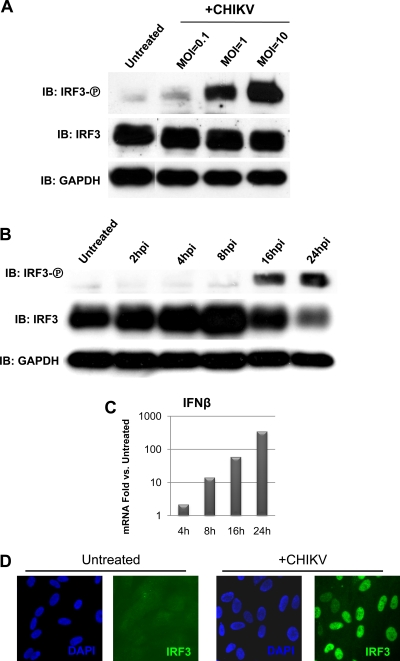
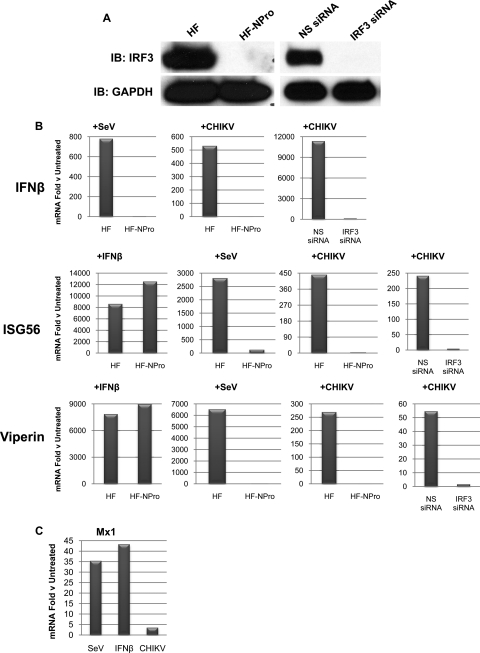
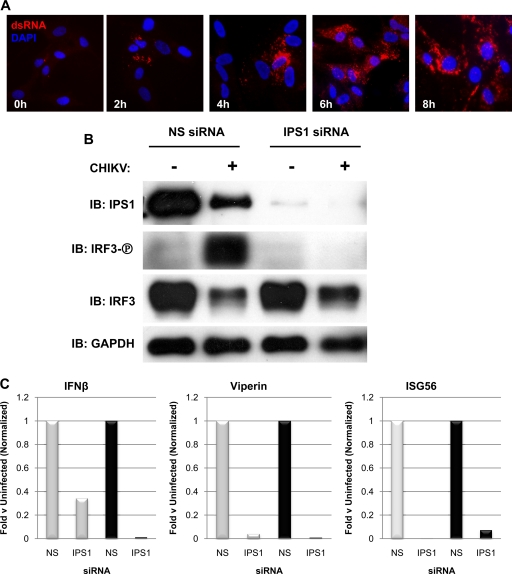
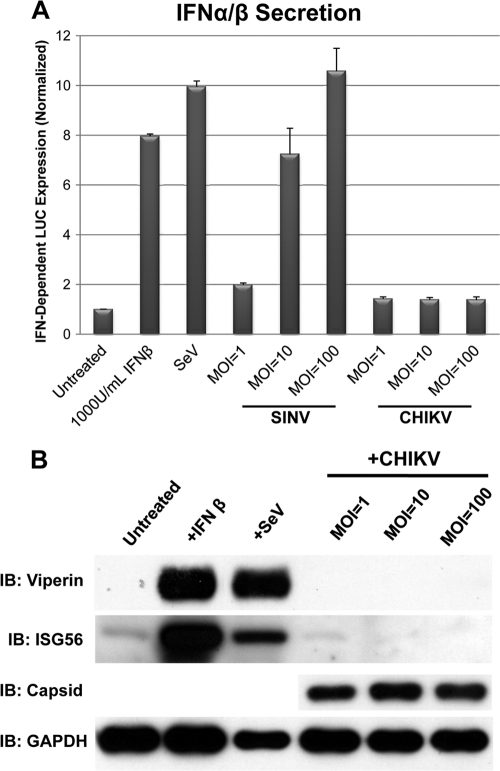
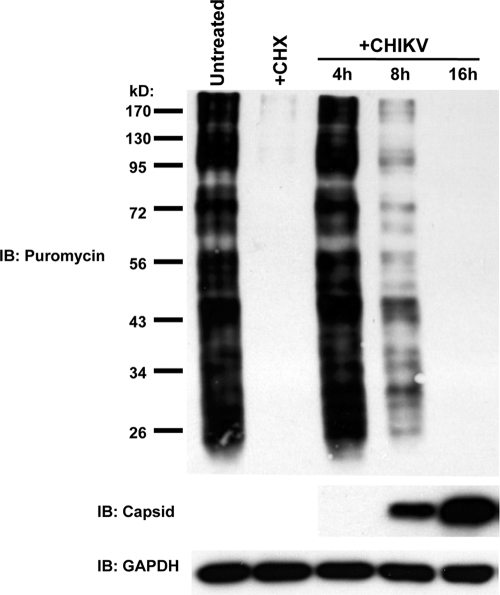
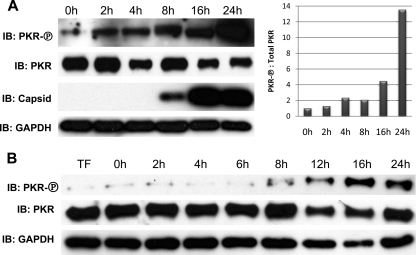
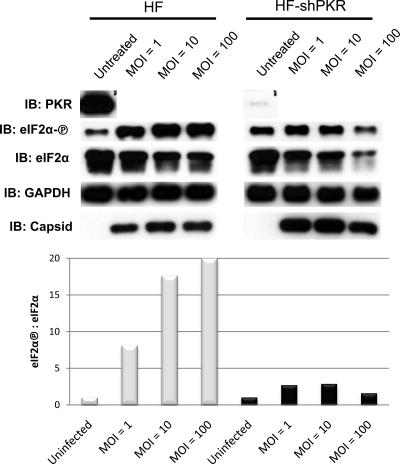
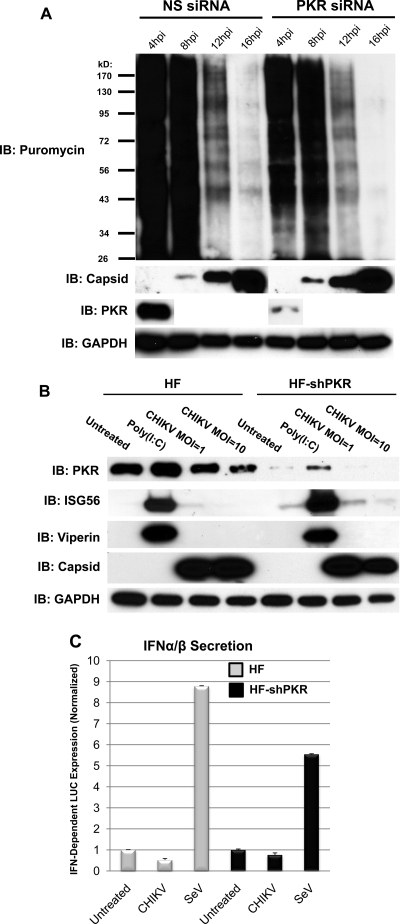
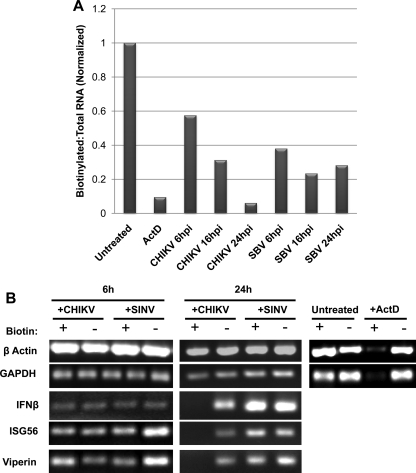
Chikungunya virus (CHIKV) is an arthritogenic mosquito-transmitted alphavirus that is undergoing reemergence in areas around the Indian Ocean. Despite the current and potential danger posed by this virus, we know surprisingly little about the induction and evasion of CHIKV-associated antiviral immune responses. With this in mind we investigated innate immune reactions to CHIKV in human fibroblasts, a demonstrable in vivo target of virus replication and spread. We show that CHIKV infection leads to activation of the transcription factor interferon regulatory factor 3 (IRF3) and subsequent transcription of IRF3-dependent antiviral genes, including beta interferon (IFN-β). IRF3 activation occurs by way of a virus-induced innate immune signaling pathway that includes the adaptor molecule interferon promoter stimulator 1 (IPS-1). Despite strong transcriptional upregulation of these genes, however, translation of the corresponding proteins is not observed. We further demonstrate that translation of cellular (but not viral) genes is blocked during infection and that although CHIKV is found to trigger inactivation of the translational molecule eukaryotic initiation factor subunit 2α by way of the double-stranded RNA sensor protein kinase R, this response is not required for the block to protein synthesis. Furthermore, overall diminution of cellular RNA synthesis is also observed in the presence of CHIKV and transcription of IRF3-dependent antiviral genes appears specifically blocked late in infection. We hypothesize that the observed absence of IFN-β and antiviral proteins during infection results from an evasion mechanism exhibited by CHIKV that is dependent on widespread shutoff of cellular protein synthesis and a targeted block to late synthesis of antiviral mRNA transcripts.
Figures
References
-
- Anishchenko, M., S. Paessler, I. P. Greene, P. V. Aguilar, A. S. Carrara, and S. C. Weaver. 2004. Generation and characterization of closely related epizootic and enzootic infectious cDNA clones for studying interferon sensitivity and emergence mechanisms of Venezuelan equine encephalitis virus. J. Virol. 78:1-8. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
