

Treating viral exacerbations of chronic obstructive pulmonary disease: insights from a mouse model of cigarette smoke and H1N1 influenza infection
- PMID: 20967263
- PMCID: PMC2953496
- DOI: 10.1371/journal.pone.0013251
Treating viral exacerbations of chronic obstructive pulmonary disease: insights from a mouse model of cigarette smoke and H1N1 influenza infection
Abstract
Background: Chronic obstructive pulmonary disease is a progressive lung disease that is punctuated by periods of exacerbations (worsening of symptoms) that are attributable to viral infections. While rhinoviruses are most commonly isolated viruses during episodes of exacerbation, influenza viruses have the potential to become even more problematic with the increased likelihood of an epidemic.
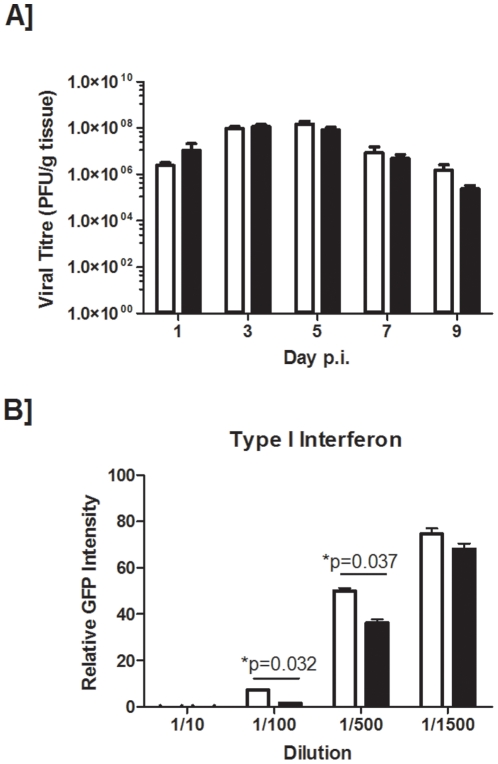
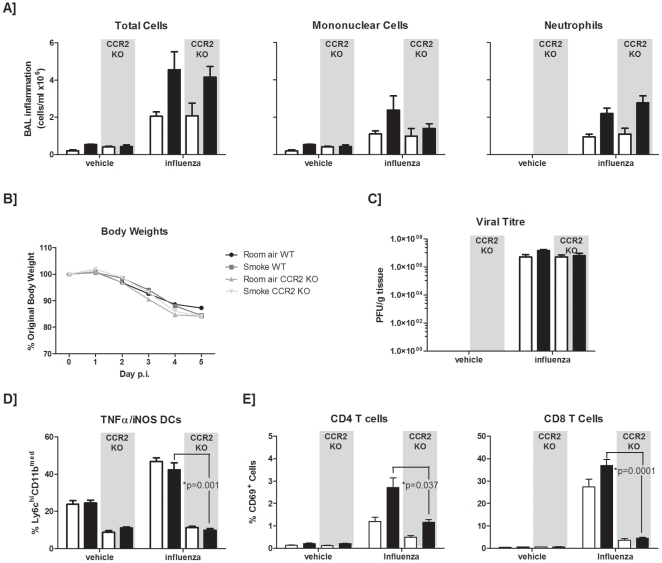
Methodology and principal findings: This study examined the impact of current and potential pharmacological targets namely the systemic corticosteroid dexamethasone and the peroxisome proliferator-activated receptor-gamma agonist pioglitazone on the outcome of infection in smoke-exposed mice. C57BL/6 mice were exposed to room air or cigarette smoke for 4 days and subsequently inoculated with an H1N1 influenza A virus. Interventions were delivered daily during the course of infection. We show that smoke-exposed mice have an exacerbated inflammatory response following infection. While smoke exposure did not compromise viral clearance, precision cut lung slices from smoke-exposed mice showed greater expression of CC (MCP-1, -3), and CXC (KC, MIP-2, GCP-2) chemokines compared to controls when stimulated with a viral mimic or influenza A virus. While dexamethasone treatment partially attenuated the inflammatory response in the broncho-alveolar lavage of smoke-exposed, virally-infected animals, viral-induced neutrophilia was steroid insensitive. In contrast to controls, dexamethasone-treated smoke-exposed influenza-infected mice had a worsened health status. Pioglitazone treatment of virally-infected smoke-exposed mice proved more efficacious than the steroid intervention. Further mechanistic evaluation revealed that a deficiency in CCR2 did not improve the inflammatory outcome in smoke-exposed, virally-infected animals.
Conclusions and significance: This animal model of cigarette smoke and H1N1 influenza infection demonstrates that smoke-exposed animals are differentially primed to respond to viral insult. While providing a platform to test pharmacological interventions, this model demonstrates that treating viral exacerbations with alternative anti-inflammatory drugs, such as PPAR-gamma agonists should be further explored since they showed greater efficacy than systemic corticosteroids.
Conflict of interest statement
Figures




References
-
- Rodriguez-Roisin R. Toward a consensus definition for COPD exacerbations. Chest. 2000;117:398S–401S. - PubMed
-
- Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. Lancet. 2007;370:765–773. - PubMed
-
- Wedzicha JA. Airway infection accelerates decline of lung function in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;164:1757–1758. - PubMed
-
- Mallia P, Johnston SL. Mechanisms and experimental models of chronic obstructive pulmonary disease exacerbations. Proc Am Thorac Soc. 2005;2:361–366; discussion 371–362. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
