

PKA and Epac synergistically inhibit smooth muscle cell proliferation
- PMID: 20971121
- PMCID: PMC3093616
- DOI: 10.1016/j.yjmcc.2010.10.010
PKA and Epac synergistically inhibit smooth muscle cell proliferation
Abstract
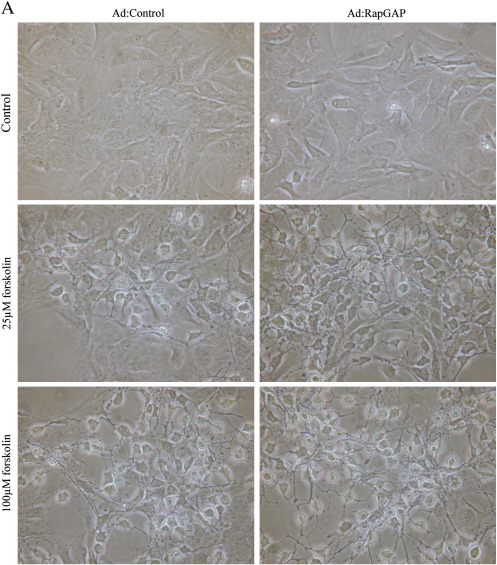
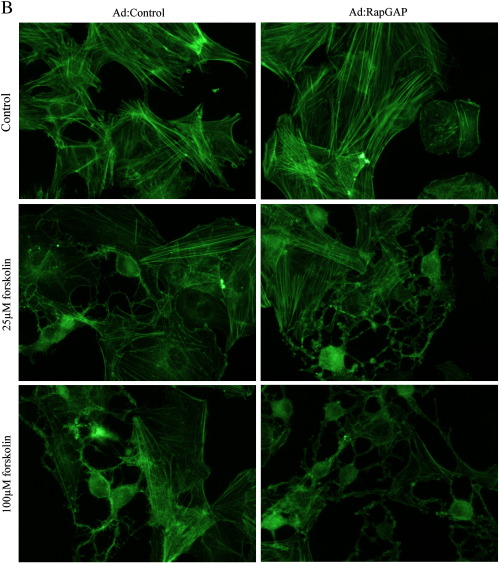
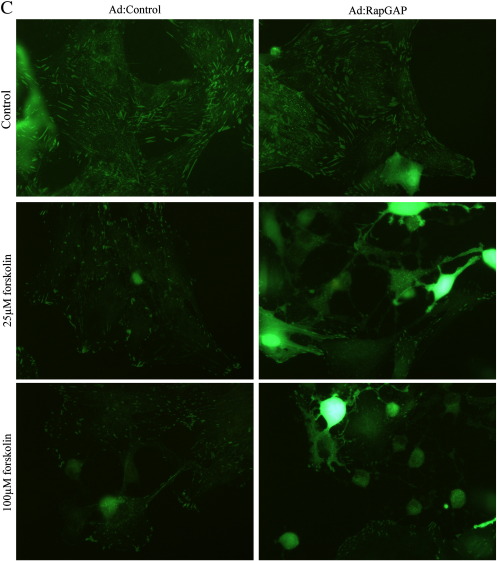
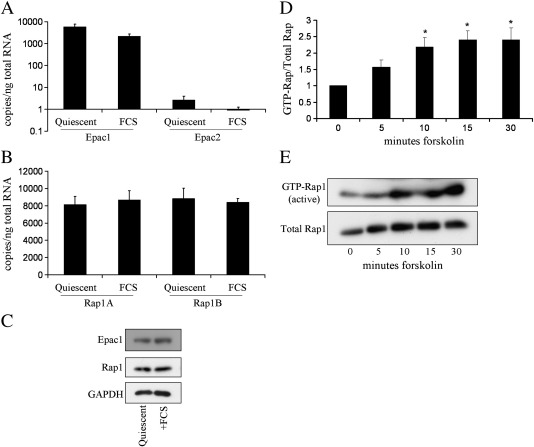
Cyclic AMP signalling promotes VSMC quiescence in healthy vessels and during vascular healing following injury. Cyclic AMP inhibits VSMC proliferation via mechanisms that are not fully understood. We investigated the role of PKA and Epac signalling on cAMP-induced inhibition of VSMC proliferation. cAMP-mediated growth arrest was PKA-dependent. However, selective PKA activation with 6-Benzoyl-cAMP did not inhibit VSMC proliferation, indicating a requirement for additional pathways. Epac activation using the selective cAMP analogue 8-CPT-2'-O-Me-cAMP, did not affect levels of hyperphosphorylated Retinoblastoma (Rb) protein, a marker of G1-S phase transition, or BrdU incorporation, despite activation of the Epac-effector Rap1. However, 6-Benzoyl-cAMP and 8-CPT-2'-O-Me-cAMP acted synergistically to inhibit Rb-hyperphosphorylation and BrdU incorporation, indicating that both pathways are required for growth inhibition. Consistent with this, constitutively active Epac increased Rap1 activity and synergised with 6-Benzoyl-cAMP to inhibit VSMC proliferation. PKA and Epac synergised to inhibit phosphorylation of ERK and JNK. Induction of stellate morphology, previously associated with cAMP-mediated growth arrest, was also dependent on activation of both PKA and Epac. Rap1 inhibition with Rap1GAP or siRNA silencing did not negate forskolin-induced inhibition of Rb-hyperphosphorylation, BrdU incorporation or stellate morphology. This data demonstrates for the first time that Epac synergises with PKA via a Rap1-independent mechanism to mediate cAMP-induced growth arrest in VSMC. This work highlights the role of Epac as a major player in cAMP-dependent growth arrest in VSMC.
Copyright © 2010 Elsevier Ltd. All rights reserved.
Figures


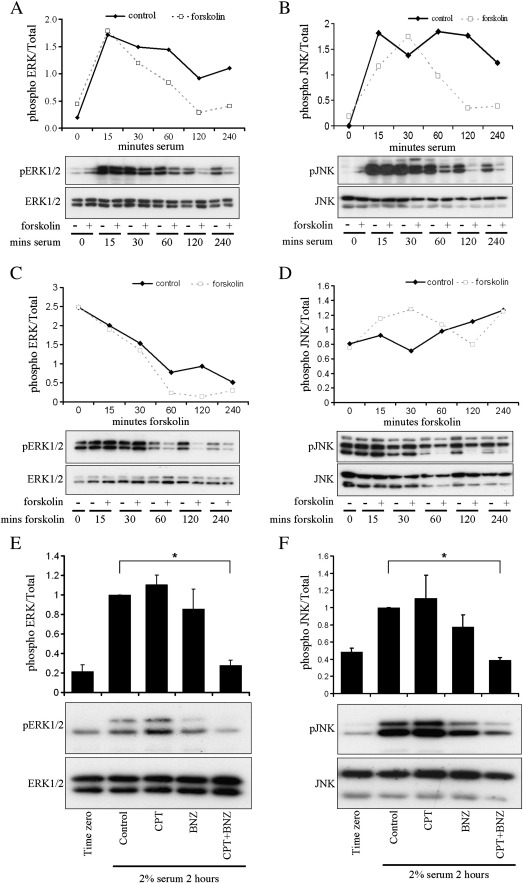
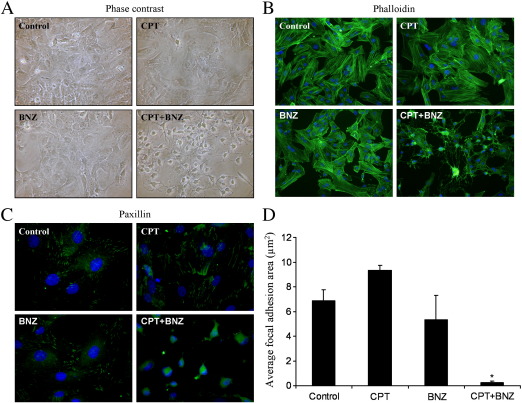
References
-
- Izzard T., Taylor C., Birkett S., Jackson C., Newby A. Mechanisms underlying maintenance of smooth muscle cell quiescence in rat aorta: role of the cyclin dependent kinases and their inhibitors. Cardiovasc Res. 2002;52:242–252. - PubMed
-
- Lewis C.D., Olson N.E., Raines E.W., Reidy M.A., Jackson C.L. Modulation of smooth muscle proliferation in rat carotid artery by platelet-derived mediators and fibroblast growth factor-2. Platelets. 2001;12:352–353. - PubMed
-
- Hedin U., Roy J., Tran P.K. Control of smooth muscle cell proliferation in vascular disease. Curr Opin Cell Biol. 2004;15:559–565. - PubMed
-
- Wu Y., Bond M., Sala-Newby G., Newby A. Altered S-phase kinase-associated protein-2 levels are a major mediator of cyclic nucleotide-induced inhibition of vascular smooth muscle cell proliferation. Circ Res. 2006;98:1141–1150. - PubMed
-
- Indolfi C., Avvedimento E.V., DiLorenzo E., Esposito G., Rapacciuolo A., Giuliano P. Activation of cAMP-PKA signaling in vivo inhibits smooth muscle cell proliferation induced by vascular injury. Nat Med. 1997;3:775–779. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
