

Protein kinases: evolution of dynamic regulatory proteins
- PMID: 20971646
- PMCID: PMC3084033
- DOI: 10.1016/j.tibs.2010.09.006
Protein kinases: evolution of dynamic regulatory proteins
Abstract
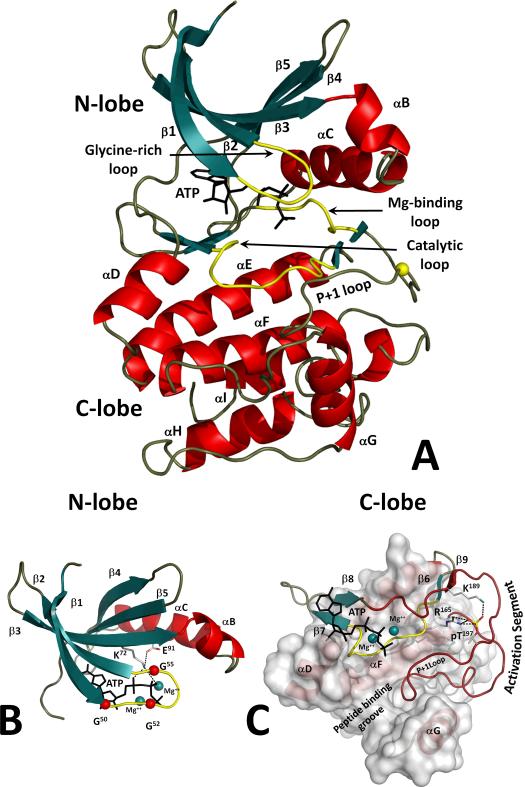
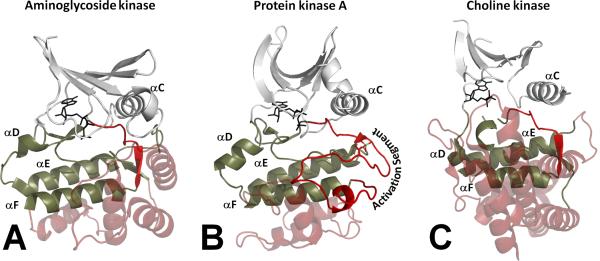
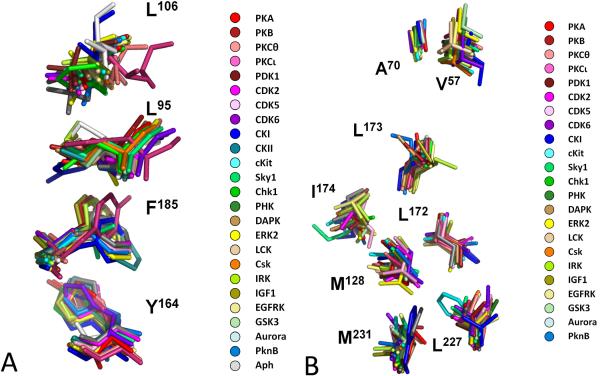
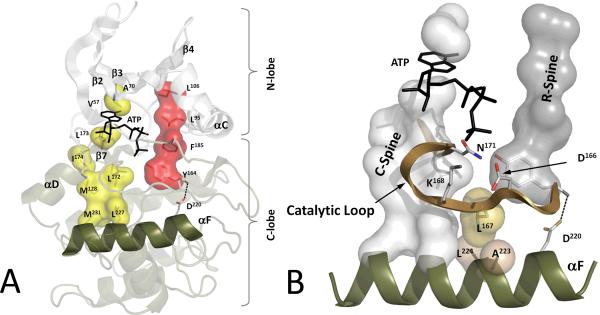
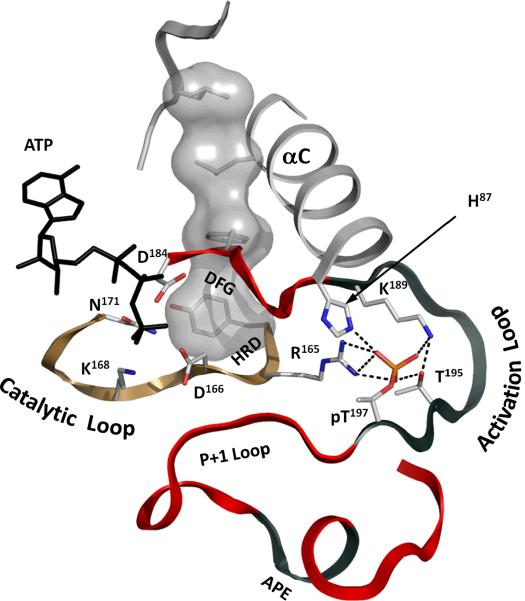
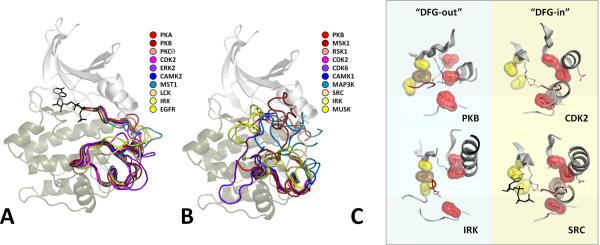
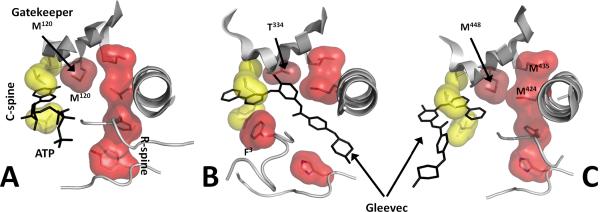
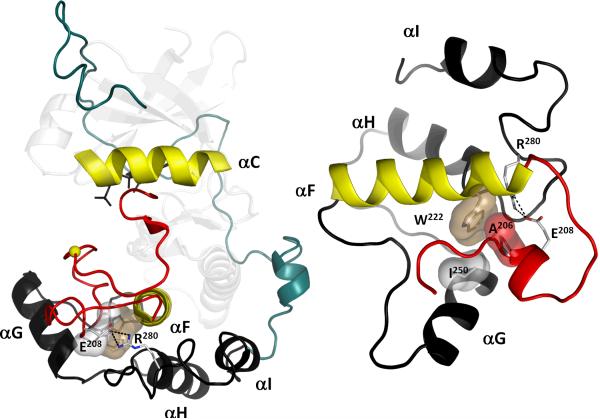
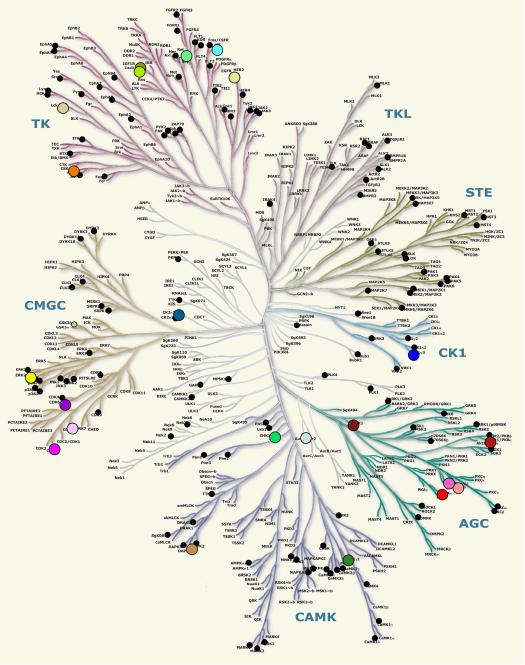
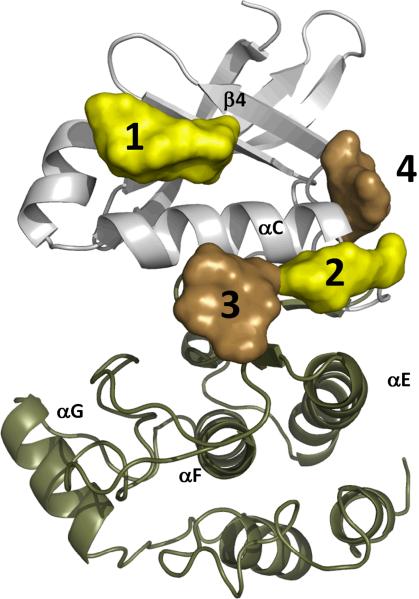
Eukayotic protein kinases evolved as a family of highly dynamic molecules with strictly organized internal architecture. A single hydrophobic F-helix serves as a central scaffold for assembly of the entire molecule. Two non-consecutive hydrophobic structures termed "spines" anchor all the elements important for catalysis to the F-helix. They make firm, but flexible, connections within the molecule, providing a high level of internal dynamics of the protein kinase. During the course of evolution, protein kinases developed a universal regulatory mechanism associated with a large activation segment that can be dynamically folded and unfolded in the course of cell functioning. Protein kinases thus represent a unique, highly dynamic, and precisely regulated set of switches that control most biological events in eukaryotic cells.
Copyright © 2010. Published by Elsevier Ltd.
Figures
References
-
- Krebs EG. An accidental biochemist. Annu Rev Biochem. 1998;67:xii–xxxii. - PubMed
-
- Walsh DA, Perkins JP, Krebs EG. An adenosine 3',5'-monophosphate-dependant protein kinase from rabbit skeletal muscle. J Biol Chem. 1968;243(13):3763–5. - PubMed
-
- Hanks SK, Hunter T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. Faseb J. 1995;9(8):576–96. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
