

Amyloid-beta protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity
- PMID: 20971854
- PMCID: PMC3030372
- DOI: 10.1074/jbc.M110.135244
Amyloid-beta protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity
Abstract
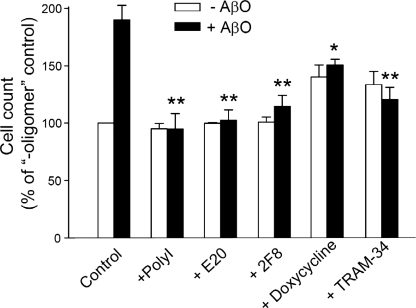
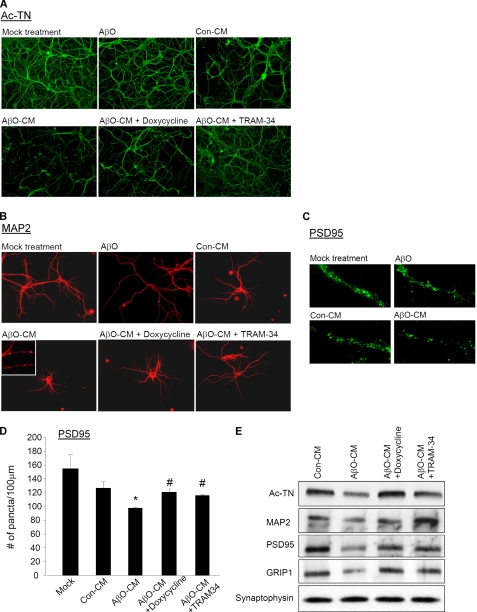
Neuroinflammation and associated neuronal dysfunction mediated by activated microglia play an important role in the pathogenesis of Alzheimer disease (AD). Microglia are activated by aggregated forms of amyloid-β protein (Aβ), usually demonstrated in vitro by stimulating microglia with micromolar concentrations of fibrillar Aβ, a major component of amyloid plaques in AD brains. Here we report that amyloid-β oligomer (AβO), at 5-50 nm, induces a unique pattern of microglia activation that requires the activity of the scavenger receptor A and the Ca(2+)-activated potassium channel KCa3.1. AβO treatment induced an activated morphological and biochemical profile of microglia, including activation of p38 MAPK and nuclear factor κB. Interestingly, although increasing nitric oxide (NO) production, AβO did not increase several proinflammatory mediators commonly induced by lipopolyliposaccharides or fibrillar Aβ, suggesting that AβO stimulates both common and divergent pathways of microglia activation. AβO at low nanomolar concentrations, although not neurotoxic, induced indirect, microglia-mediated damage to neurons in dissociated cultures and in organotypic hippocampal slices. The indirect neurotoxicity was prevented by (i) doxycycline, an inhibitor of microglia activation; (ii) TRAM-34, a selective KCa3.1 blocker; and (iii) two inhibitors of inducible NO synthase, indicating that KCa3.1 activity and excessive NO release are required for AβO-induced microglial neurotoxicity. Our results suggest that AβO, generally considered a neurotoxin, may more potently cause neuronal damage indirectly by activating microglia in AD.
Figures






Similar articles
-
Donepezil inhibits the amyloid-beta oligomer-induced microglial activation in vitro and in vivo.Neurotoxicology. 2014 Jan;40:23-32. doi: 10.1016/j.neuro.2013.10.004. Epub 2013 Nov 1. Neurotoxicology. 2014. PMID: 24189446
-
Repurposing the KCa3.1 inhibitor senicapoc for Alzheimer's disease.Ann Clin Transl Neurol. 2019 Mar 18;6(4):723-738. doi: 10.1002/acn3.754. eCollection 2019 Apr. Ann Clin Transl Neurol. 2019. PMID: 31019997 Free PMC article.
-
Adiponectin suppresses amyloid-β oligomer (AβO)-induced inflammatory response of microglia via AdipoR1-AMPK-NF-κB signaling pathway.J Neuroinflammation. 2019 May 25;16(1):110. doi: 10.1186/s12974-019-1492-6. J Neuroinflammation. 2019. PMID: 31128596 Free PMC article.
-
Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer's disease.Int J Neurosci. 2014 May;124(5):307-21. doi: 10.3109/00207454.2013.833510. Epub 2013 Sep 12. Int J Neurosci. 2014. PMID: 23930978 Review.
-
Fibrillar beta-amyloid induces microglial phagocytosis, expression of inducible nitric oxide synthase, and loss of a select population of neurons in the rat CNS in vivo.J Neurosci. 1998 Mar 15;18(6):2161-73. doi: 10.1523/JNEUROSCI.18-06-02161.1998. J Neurosci. 1998. PMID: 9482801 Free PMC article. Review.
Cited by
-
Amyloid-β immunization enhances neurogenesis and cognitive ability in neonatal mice.Int J Clin Exp Med. 2015 Apr 15;8(4):5340-50. eCollection 2015. Int J Clin Exp Med. 2015. PMID: 26131110 Free PMC article.
-
Oligomeric Aβ-induced microglial activation is possibly mediated by NADPH oxidase.Neurochem Res. 2013 Feb;38(2):443-52. doi: 10.1007/s11064-012-0939-2. Epub 2012 Dec 11. Neurochem Res. 2013. PMID: 23229789
-
Licochalcone E, a β-Amyloid Aggregation Inhibitor, Regulates Microglial M1/M2 Polarization via Inhibition of CTL1-Mediated Choline Uptake.Biomolecules. 2023 Jan 17;13(2):191. doi: 10.3390/biom13020191. Biomolecules. 2023. PMID: 36830561 Free PMC article.
-
Microglia in Alzheimer's Disease: A Role for Ion Channels.Front Neurosci. 2018 Sep 28;12:676. doi: 10.3389/fnins.2018.00676. eCollection 2018. Front Neurosci. 2018. PMID: 30323735 Free PMC article. Review.
-
Deoxyglucose prevents neurodegeneration in culture by eliminating microglia.J Neuroinflammation. 2014 Mar 26;11:58. doi: 10.1186/1742-2094-11-58. J Neuroinflammation. 2014. PMID: 24669778 Free PMC article.
References
-
- Terry R. D., Masliah E., Salmon D. P., Butters N., DeTeresa R., Hill R., Hansen L. A., Katzman R. (1991) Ann. Neurol. 30, 572–580 - PubMed
-
- Hardy J., Selkoe D. J. (2002) Science 297, 353–356 - PubMed
-
- Edison P., Archer H. A., Gerhard A., Hinz R., Pavese N., Turkheimer F. E., Hammers A., Tai Y. F., Fox N., Kennedy A., Rossor M., Brooks D. J. (2008) Neurobiol. Dis. 32, 412–419 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
