

Child health, developmental plasticity, and epigenetic programming
- PMID: 20971919
- PMCID: PMC3365792
- DOI: 10.1210/er.2009-0039
Child health, developmental plasticity, and epigenetic programming
Abstract
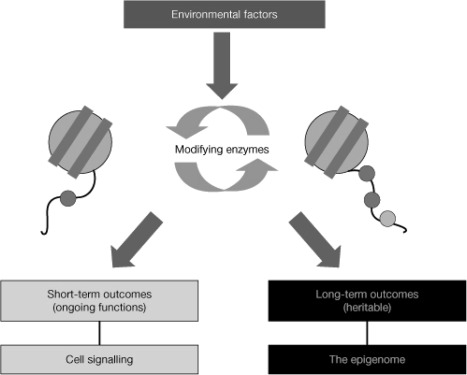
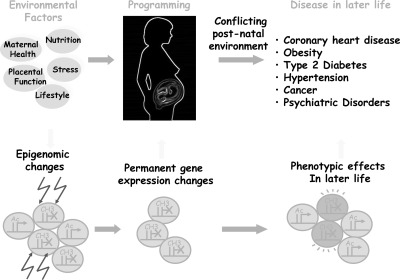
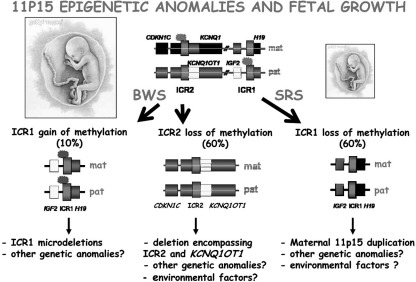
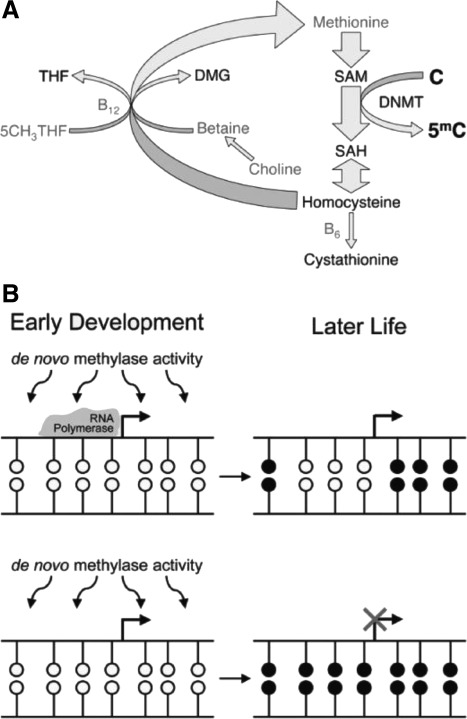
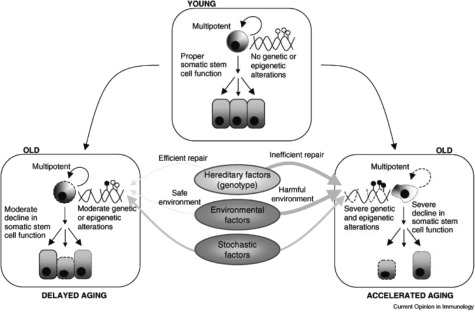
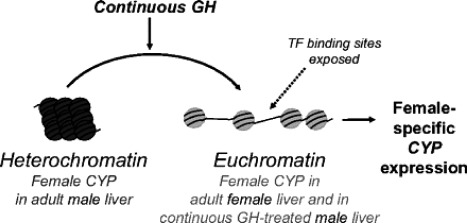
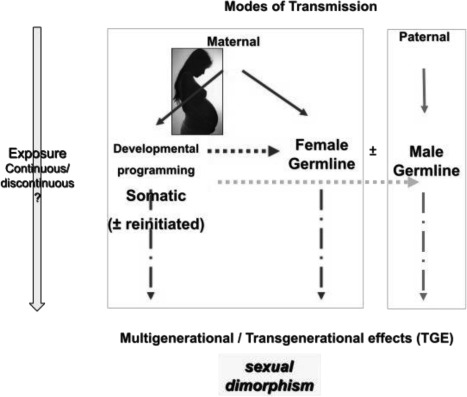
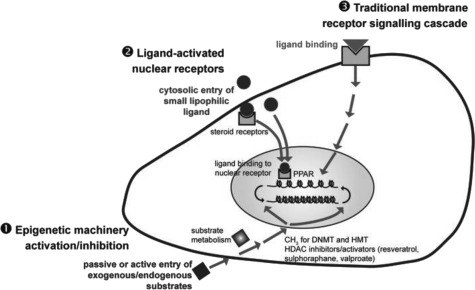
Plasticity in developmental programming has evolved in order to provide the best chances of survival and reproductive success to the organism under changing environments. Environmental conditions that are experienced in early life can profoundly influence human biology and long-term health. Developmental origins of health and disease and life-history transitions are purported to use placental, nutritional, and endocrine cues for setting long-term biological, mental, and behavioral strategies in response to local ecological and/or social conditions. The window of developmental plasticity extends from preconception to early childhood and involves epigenetic responses to environmental changes, which exert their effects during life-history phase transitions. These epigenetic responses influence development, cell- and tissue-specific gene expression, and sexual dimorphism, and, in exceptional cases, could be transmitted transgenerationally. Translational epigenetic research in child health is a reiterative process that ranges from research in the basic sciences, preclinical research, and pediatric clinical research. Identifying the epigenetic consequences of fetal programming creates potential applications in clinical practice: the development of epigenetic biomarkers for early diagnosis of disease, the ability to identify susceptible individuals at risk for adult diseases, and the development of novel preventive and curative measures that are based on diet and/or novel epigenetic drugs.
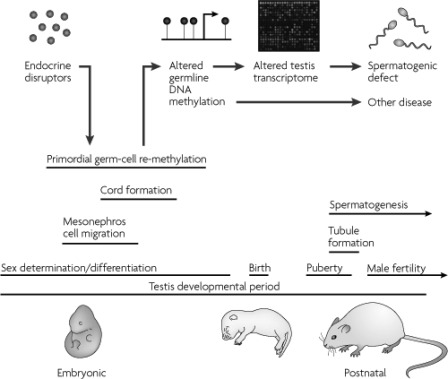
Figures





References
-
- Bateson P. 2005. The return of the whole organism. J Biosci 30:31–39 - PubMed
-
- Crews D, McLachlan JA. 2006. Epigenetics, evolution, endocrine disruption, health, and disease. Endocrinology 147:S4–S10 - PubMed
-
- Hochberg Z. 2009. Evo-devo of child growth II: human life history and transition between its phases. Eur J Endocrinol 160:135–141 - PubMed
-
- Barker DJ. 1992. The fetal origins of adult hypertension. J Hypertens 10:S39–S44 - PubMed
