

A novel form of human disease with a protease-sensitive prion protein and heterozygosity methionine/valine at codon 129: Case report
- PMID: 20973975
- PMCID: PMC2987858
- DOI: 10.1186/1471-2377-10-99
A novel form of human disease with a protease-sensitive prion protein and heterozygosity methionine/valine at codon 129: Case report
Abstract
Background: Sporadic Creutzfeldt-Jakob disease (sCJD) is a rare neurodegenerative disorder in humans included in the group of Transmissible Spongiform Encephalopathies or prion diseases. The vast majority of sCJD cases are molecularly classified according to the abnormal prion protein (PrPSc) conformations along with polymorphism of codon 129 of the PRNP gene. Recently, a novel human disease, termed "protease-sensitive prionopathy", has been described. This disease shows a distinct clinical and neuropathological phenotype and it is associated to an abnormal prion protein more sensitive to protease digestion.
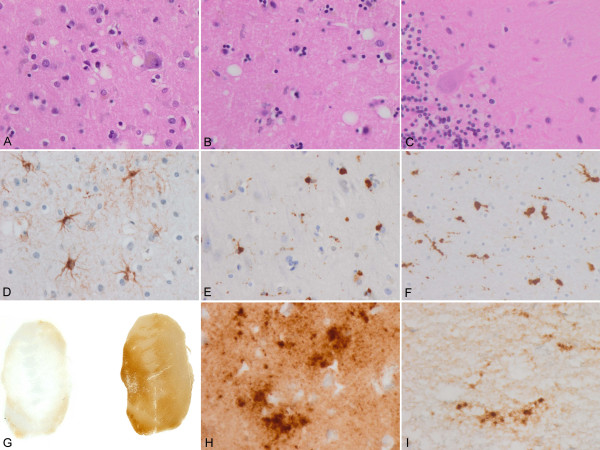
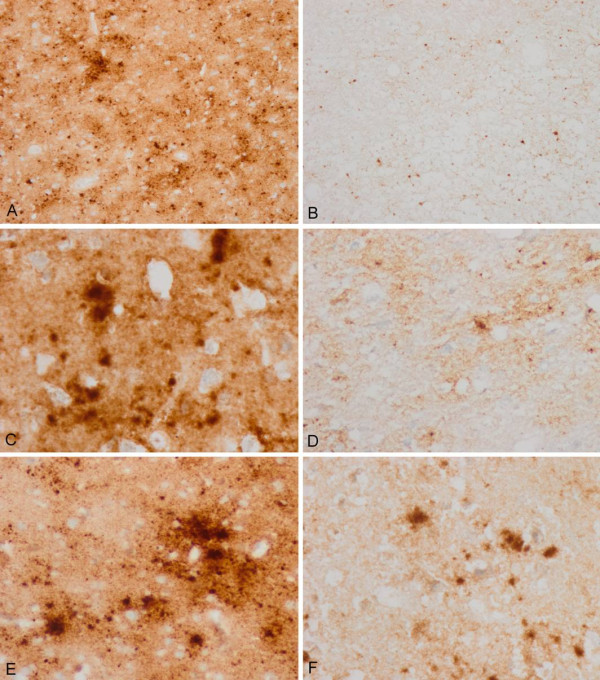
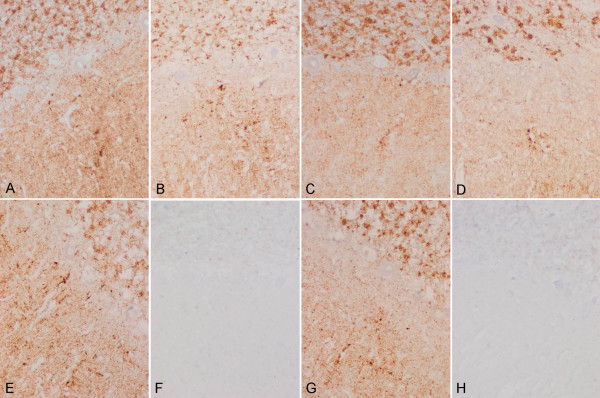
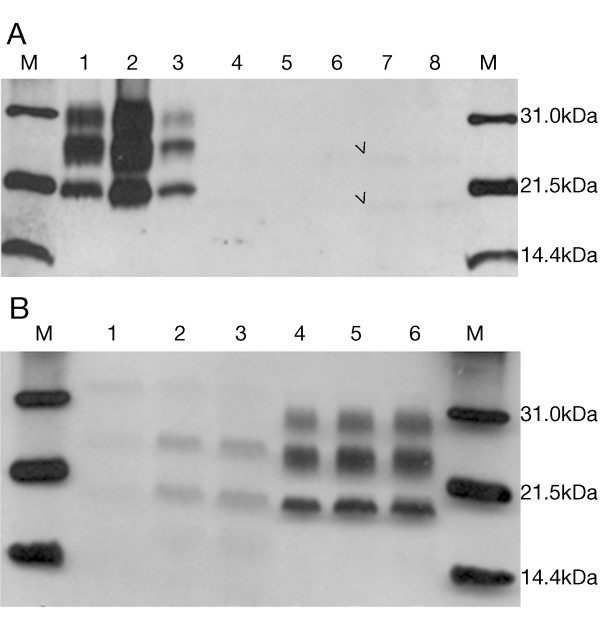
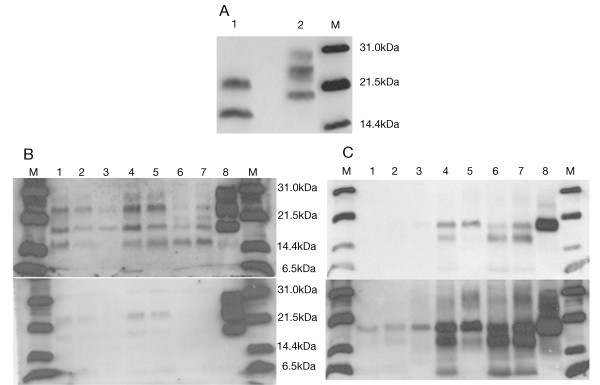
Case presentation: We report the case of a 75-year-old-man who developed a clinical course and presented pathologic lesions compatible with sporadic Creutzfeldt-Jakob disease, and biochemical findings reminiscent of "protease-sensitive prionopathy". Neuropathological examinations revealed spongiform change mainly affecting the cerebral cortex, putamen/globus pallidus and thalamus, accompanied by mild astrocytosis and microgliosis, with slight involvement of the cerebellum. Confluent vacuoles were absent. Diffuse synaptic PrP deposits in these regions were largely removed following proteinase treatment. PrP deposition, as revealed with 3F4 and 1E4 antibodies, was markedly sensitive to pre-treatment with proteinase K. Molecular analysis of PrPSc showed an abnormal prion protein more sensitive to proteinase K digestion, with a five-band pattern of 28, 24, 21, 19, and 16 kDa, and three aglycosylated isoforms of 19, 16 and 6 kDa. This PrPSc was estimated to be 80% susceptible to digestion while the pathogenic prion protein associated with classical forms of sporadic Creutzfeldt-Jakob disease were only 2% (type VV2) and 23% (type MM1) susceptible. No mutations in the PRNP gene were found and genotype for codon 129 was heterozygous methionine/valine.
Conclusions: A novel form of human disease with abnormal prion protein sensitive to protease and MV at codon 129 was described. Although clinical signs were compatible with sporadic Creutzfeldt-Jakob disease, the molecular subtype with the abnormal prion protein isoforms showing enhanced protease sensitivity was reminiscent of the "protease-sensitive prionopathy". It remains to be established whether the differences found between the latter and this case are due to the polymorphism at codon 129. Different degrees of proteinase K susceptibility were easily determined with the chemical polymer detection system which could help to detect proteinase-susceptible pathologic prion protein in diseases other than the classical ones.
Figures
References
-
- Ladogana A, Puopolo M, Croes EA, Budka H, Jarius C, Collins S, Klug GM, Sutcliffe T, Giulivi A, Alperovitch A. et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology. 2005;64:1586–1591. doi: 10.1212/01.WNL.0000160117.56690.B2. - DOI - PubMed
-
- Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P. et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–233. doi: 10.1002/1531-8249(199908)46:2<224::AID-ANA12>3.0.CO;2-W. - DOI - PubMed
-
- Budka H. Histopathology and immunohistochemistry of human transmissible spongiform encephalopathies (TSEs) Arch Virol Suppl. 2000;16:135–142. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
