

AAV-mediated gene delivery in adult GM1-gangliosidosis mice corrects lysosomal storage in CNS and improves survival
- PMID: 20976108
- PMCID: PMC2956705
- DOI: 10.1371/journal.pone.0013468
AAV-mediated gene delivery in adult GM1-gangliosidosis mice corrects lysosomal storage in CNS and improves survival
Abstract
Background: GM1-gangliosidosis is a glycosphingolipid (GSL) lysosomal storage disease caused by a genetic deficiency of acid β-galactosidase (βgal), which results in the accumulation of GM1-ganglioside and its asialo-form (GA1) primarily in the CNS. Age of onset ranges from infancy to adulthood, and excessive ganglioside accumulation produces progressive neurodegeneration and psychomotor retardation in humans. Currently, there are no effective therapies for the treatment of GM1-gangliosidosis.
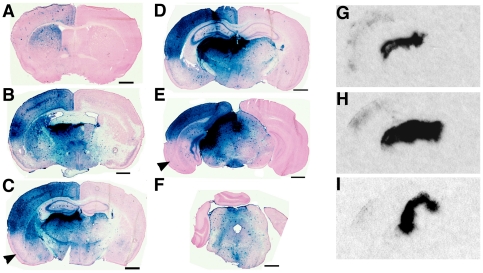
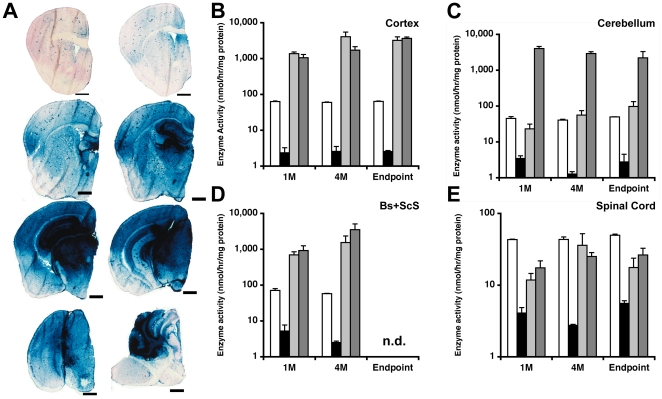
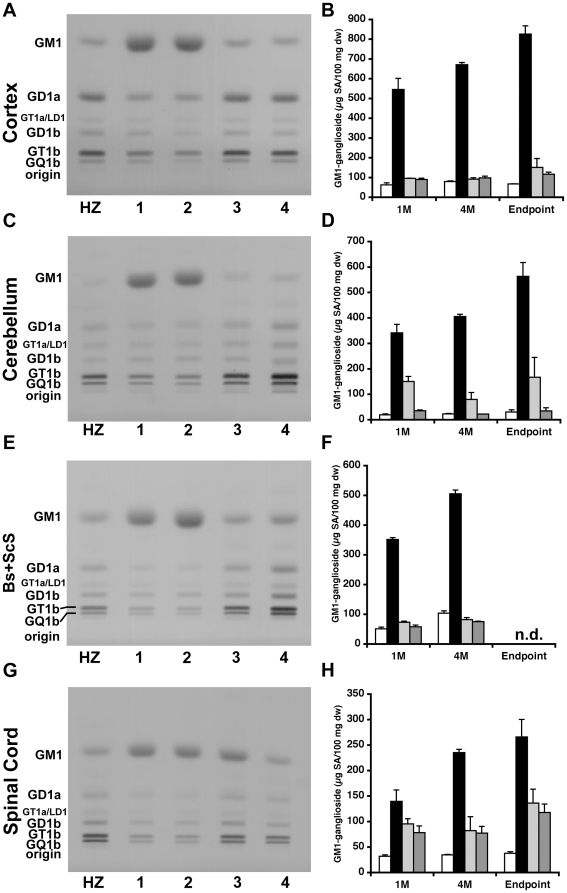
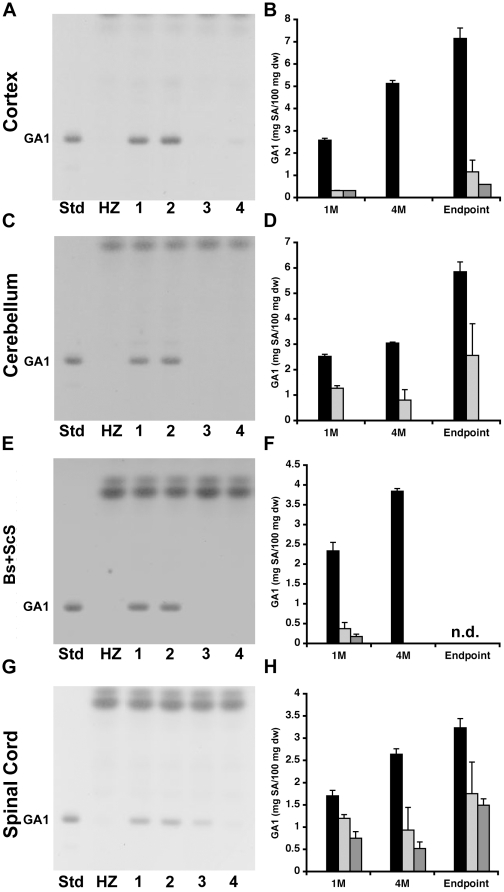
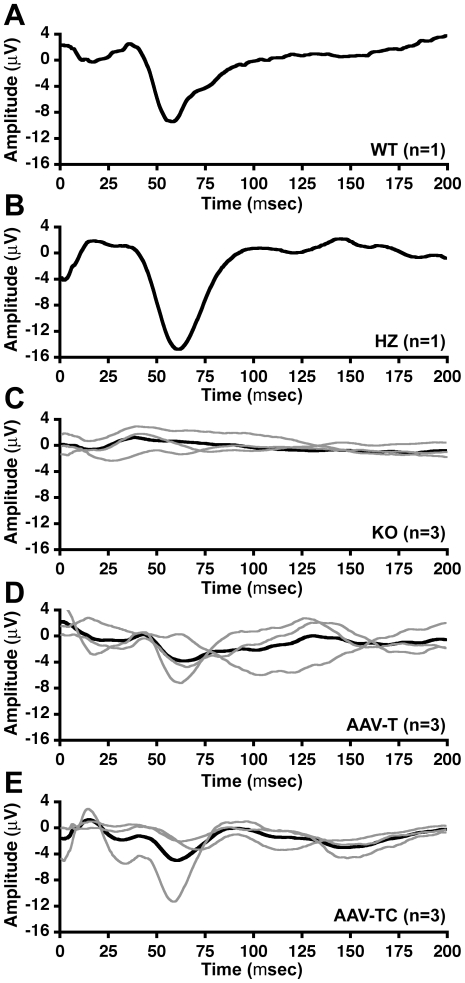
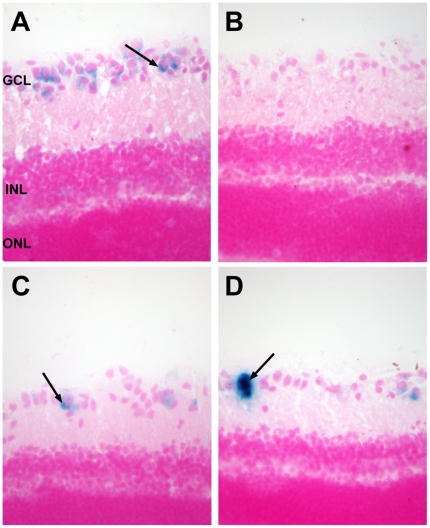
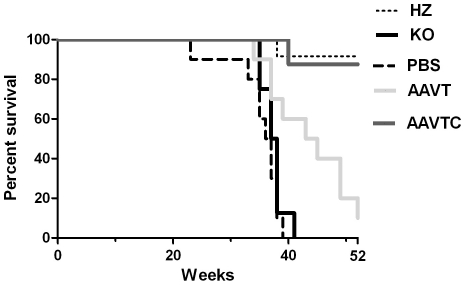
Methodology/principal findings: In this study we examined the effect of thalamic infusion of AAV2/1-βgal vector in adult GM1 mice on enzyme distribution, activity, and GSL content in the CNS, motor behavior, and survival. Six to eight week-old GM1 mice received bilateral injections of AAV vector in the thalamus, or thalamus and deep cerebellar nuclei (DCN) with pre-determined endpoints at 1 and 4 months post-injection, and the humane endpoint, or 52 weeks of age. Enzyme activity was elevated throughout the CNS of AAV-treated GM1 mice and GSL storage nearly normalized in most structures analyzed, except in the spinal cord which showed ∼50% reduction compared to age-matched untreated GM1 mice spinal cord. Survival was significantly longer in AAV-treated GM1 mice (52 wks) than in untreated mice. However the motor performance of AAV-treated GM1 mice declined over time at a rate similar to that observed in untreated GM1 mice.
Conclusions/significance: Our studies show that the AAV-modified thalamus can be used as a 'built-in' central node network for widespread distribution of lysosomal enzymes in the mouse cerebrum. In addition, this study indicates that thalamic delivery of AAV vectors should be combined with additional targets to supply the cerebellum and spinal cord with therapeutic levels of enzyme necessary to achieve complete correction of the neurological phenotype in GM1 mice.
Conflict of interest statement
Figures

References
-
- Okada S, O'Brien JS. Generalized gangliosidosis: beta-galactosidase deficiency. Science. 1968;160:1002–1004. - PubMed
-
- O'Brien JS, Stern MB, Landing BH, O'Brien JK, Donnell GN. Generalized Gangliosidosis: Another Inborn Error of Ganglioside Metabolism? Am J Dis Child. 1965;109:338–346. - PubMed
-
- Suzuki Y, Oshima A, Nanba E. β-galactosidase deficiency (β-galactosidosis): GM1 gangliosidosis and Morquio B disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Online Metabolic & Molecular Basis of Inherited Diseases. New York: McGraw-Hill; 2001. pp. 3775–3810.
-
- Bieber FR, Mortimer G, Kolodny EH, Driscoll SG. Pathologic findings in fetal GM1 gangliosidosis. Arch Neurol. 1986;43:736–738. - PubMed
-
- Kudoh T, Kikuchi K, Nakamura F, Yokoyama S, Karube K, et al. Prenatal diagnosis of GM1-gangliosidosis: biochemical manifestations in fetal tissues. Hum Genet. 1978;44:287–293. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
