

A metagenomic analysis of pandemic influenza A (2009 H1N1) infection in patients from North America
- PMID: 20976137
- PMCID: PMC2956640
- DOI: 10.1371/journal.pone.0013381
A metagenomic analysis of pandemic influenza A (2009 H1N1) infection in patients from North America
Abstract
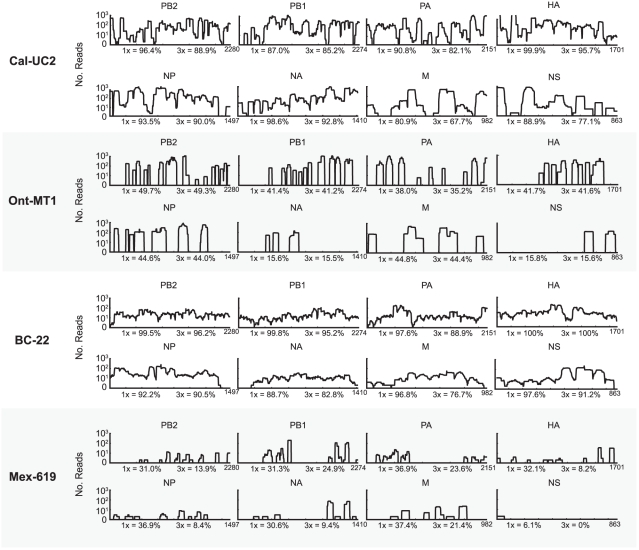
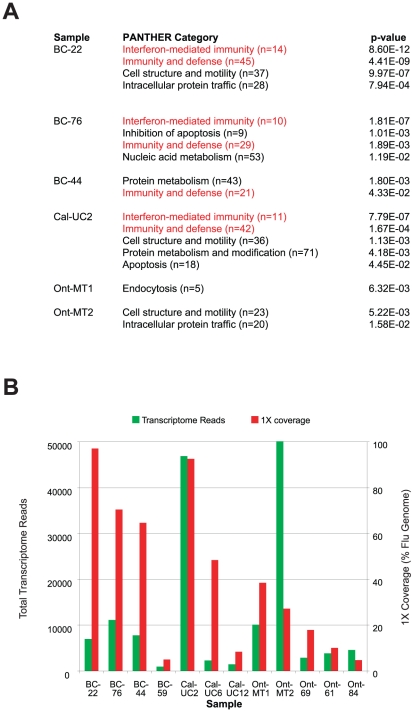
Although metagenomics has been previously employed for pathogen discovery, its cost and complexity have prevented its use as a practical front-line diagnostic for unknown infectious diseases. Here we demonstrate the utility of two metagenomics-based strategies, a pan-viral microarray (Virochip) and deep sequencing, for the identification and characterization of 2009 pandemic H1N1 influenza A virus. Using nasopharyngeal swabs collected during the earliest stages of the pandemic in Mexico, Canada, and the United States (n = 17), the Virochip was able to detect a novel virus most closely related to swine influenza viruses without a priori information. Deep sequencing yielded reads corresponding to 2009 H1N1 influenza in each sample (percentage of aligned sequences corresponding to 2009 H1N1 ranging from 0.0011% to 10.9%), with up to 97% coverage of the influenza genome in one sample. Detection of 2009 H1N1 by deep sequencing was possible even at titers near the limits of detection for specific RT-PCR, and the percentage of sequence reads was linearly correlated with virus titer. Deep sequencing also provided insights into the upper respiratory microbiota and host gene expression in response to 2009 H1N1 infection. An unbiased analysis combining sequence data from all 17 outbreak samples revealed that 90% of the 2009 H1N1 genome could be assembled de novo without the use of any reference sequence, including assembly of several near full-length genomic segments. These results indicate that a streamlined metagenomics detection strategy can potentially replace the multiple conventional diagnostic tests required to investigate an outbreak of a novel pathogen, and provide a blueprint for comprehensive diagnosis of unexplained acute illnesses or outbreaks in clinical and public health settings.
Conflict of interest statement
Figures
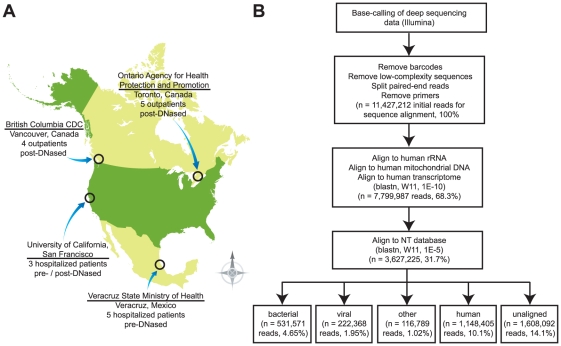
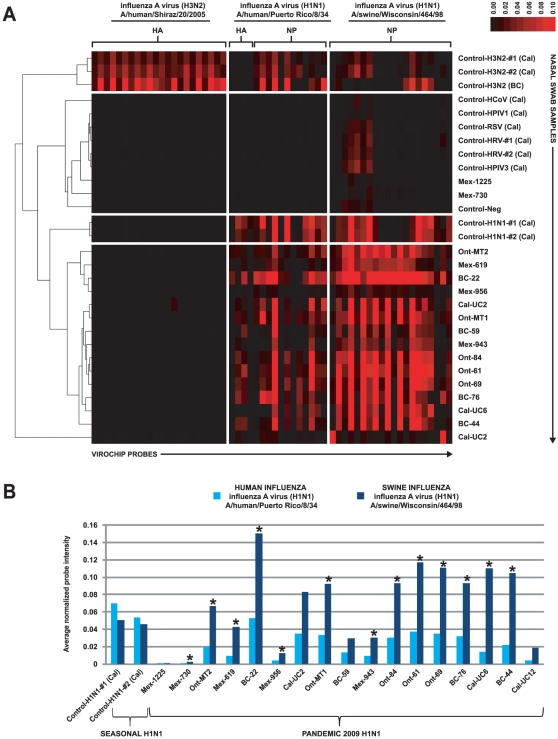
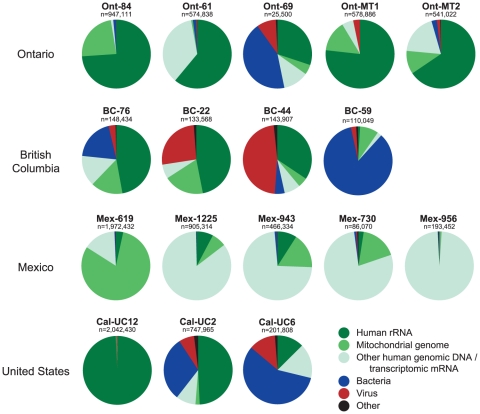
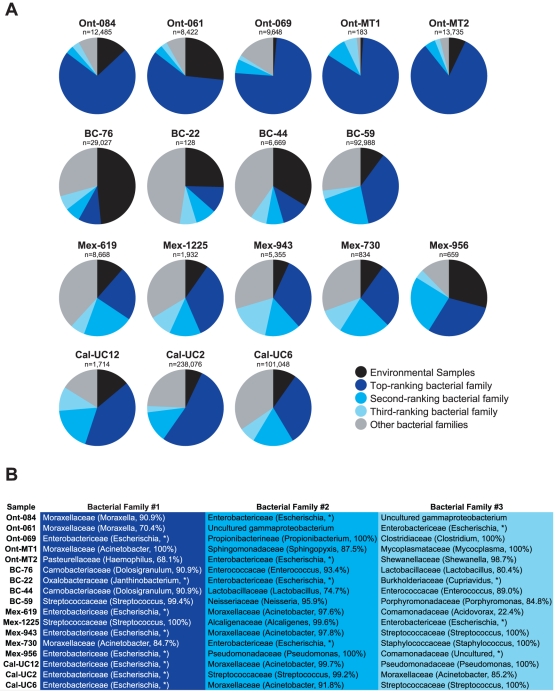


References
-
- Dawood FS, Jain S, Finelli L, Shaw MW, et al. Novel Swine-Origin Influenza AVIT. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N Engl J Med. 2009;360:2605–2615. - PubMed
-
- Shinde V, Bridges CB, Uyeki TM, Shu B, Balish A, et al. Triple-reassortant swine influenza A (H1) in humans in the United States, 2005-2009. N Engl J Med. 2009;360:2616–2625. - PubMed
-
- Faix DJ, Sherman SS, Waterman SH. Rapid-test sensitivity for novel swine-origin influenza A (H1N1) virus in humans. N Engl J Med. 2009;361:728–729. - PubMed
-
- Smith GJ, Vijaykrishna D, Bahl J, Lycett SJ, Worobey M, et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature. 2009;459:1122–1125. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
