

Characterising and predicting haploinsufficiency in the human genome
- PMID: 20976243
- PMCID: PMC2954820
- DOI: 10.1371/journal.pgen.1001154
Characterising and predicting haploinsufficiency in the human genome
Abstract
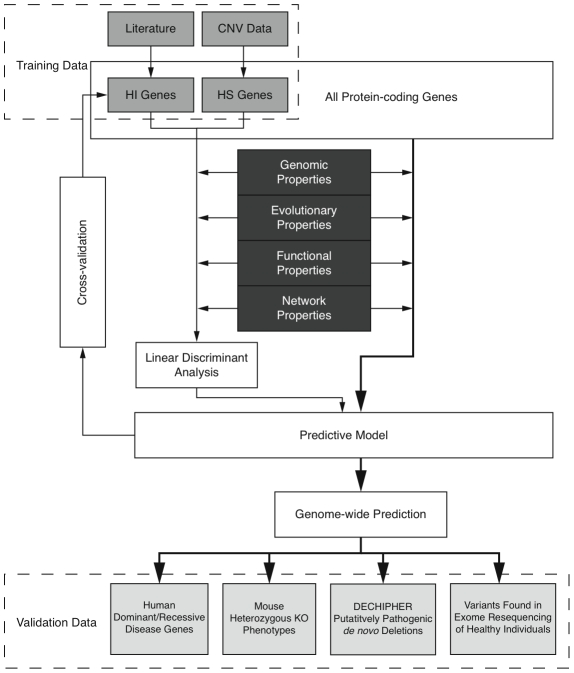
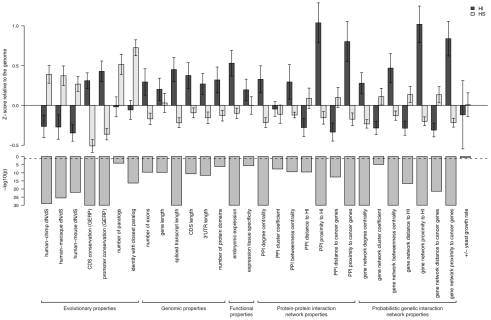
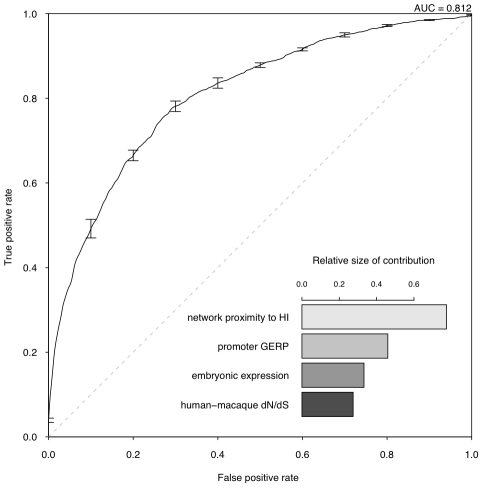
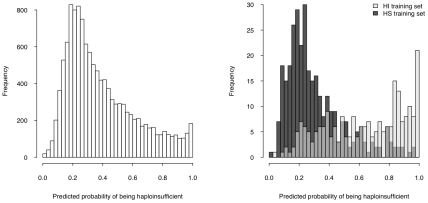
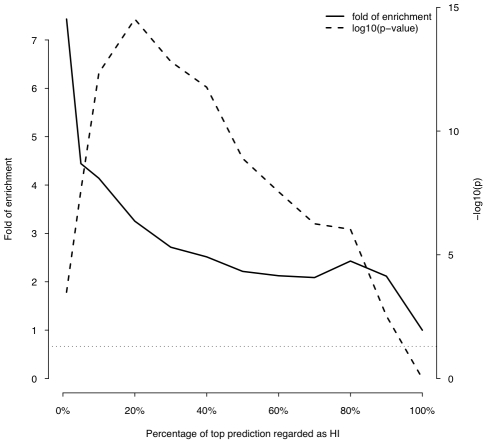
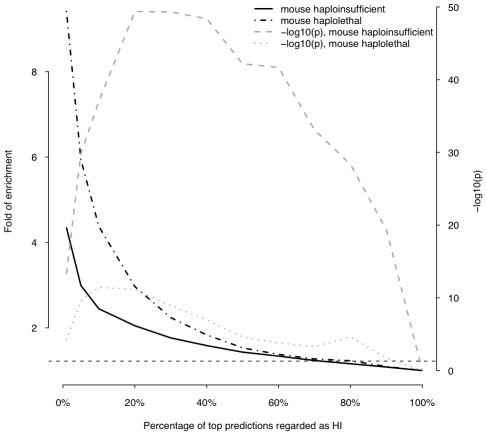
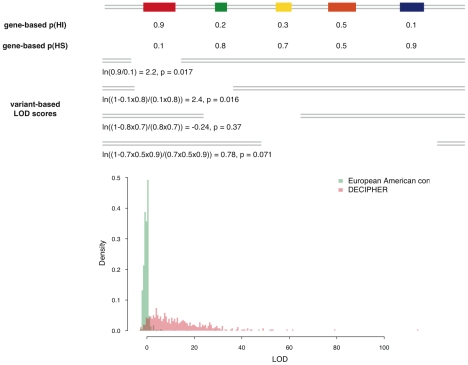
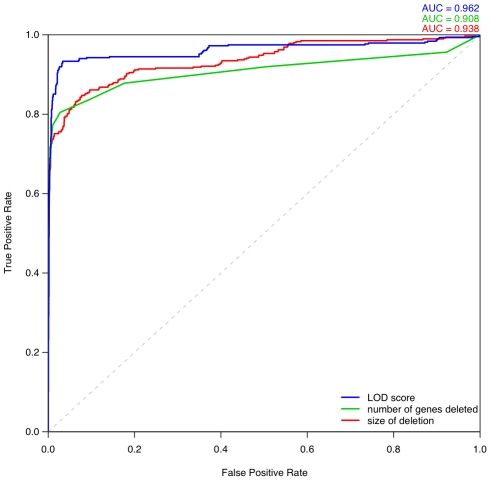
Haploinsufficiency, wherein a single functional copy of a gene is insufficient to maintain normal function, is a major cause of dominant disease. Human disease studies have identified several hundred haploinsufficient (HI) genes. We have compiled a map of 1,079 haplosufficient (HS) genes by systematic identification of genes unambiguously and repeatedly compromised by copy number variation among 8,458 apparently healthy individuals and contrasted the genomic, evolutionary, functional, and network properties between these HS genes and known HI genes. We found that HI genes are typically longer and have more conserved coding sequences and promoters than HS genes. HI genes exhibit higher levels of expression during early development and greater tissue specificity. Moreover, within a probabilistic human functional interaction network HI genes have more interaction partners and greater network proximity to other known HI genes. We built a predictive model on the basis of these differences and annotated 12,443 genes with their predicted probability of being haploinsufficient. We validated these predictions of haploinsufficiency by demonstrating that genes with a high predicted probability of exhibiting haploinsufficiency are enriched among genes implicated in human dominant diseases and among genes causing abnormal phenotypes in heterozygous knockout mice. We have transformed these gene-based haploinsufficiency predictions into haploinsufficiency scores for genic deletions, which we demonstrate to better discriminate between pathogenic and benign deletions than consideration of the deletion size or numbers of genes deleted. These robust predictions of haploinsufficiency support clinical interpretation of novel loss-of-function variants and prioritization of variants and genes for follow-up studies.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annu Rev Genomics Hum Genet. 2006;7:61–80. - PubMed
-
- Ng PC, Levy S, Huang J, Stockwell TB, Walenz BP, et al. Genetic variation in an individual human exome. PLoS Genet. 2008;4:e1000160. doi: 10.1371/journal.pgen.1000160. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
