

Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice
- PMID: 20978347
- PMCID: PMC2964987
- DOI: 10.1172/JCI43490
Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice
Abstract
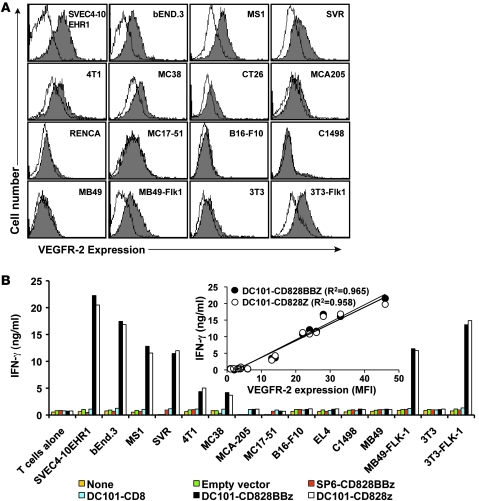
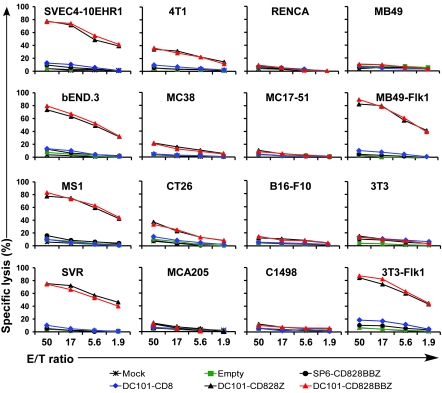
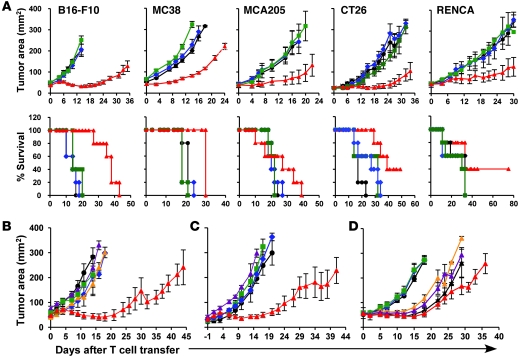
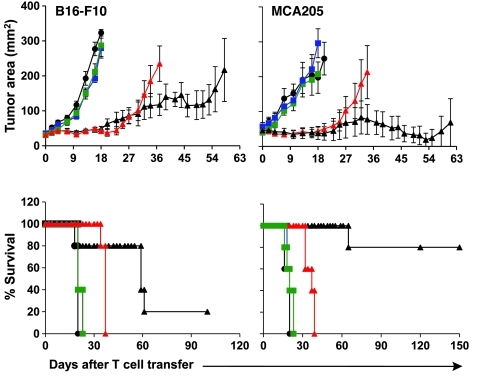
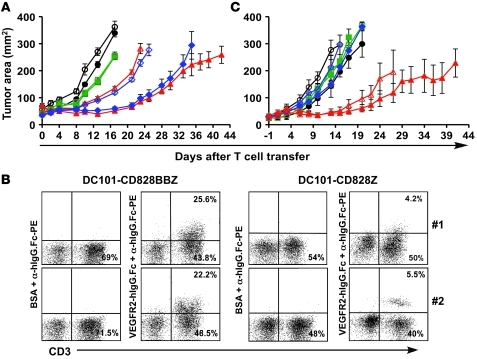
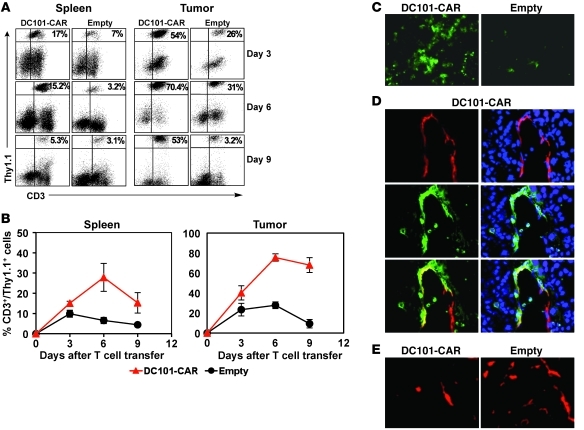
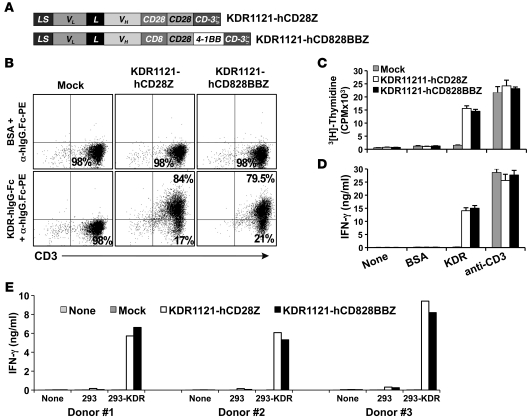
Immunotherapies based on adoptive cell transfer are highly effective in the treatment of metastatic melanoma, but the use of this approach in other cancer histologies has been hampered by the identification of appropriate target molecules. Immunologic approaches targeting tumor vasculature provide a means for the therapy of multiple solid tumor types. We developed a method to target tumor vasculature, using genetically redirected syngeneic or autologous T cells. Mouse and human T cells were engineered to express a chimeric antigen receptor (CAR) targeted against VEGFR-2, which is overexpressed in tumor vasculature and is responsible for VEGF-mediated tumor progression and metastasis. Mouse and human T cells expressing the relevant VEGFR-2 CARs mediated specific immune responses against VEGFR-2 protein as well as VEGFR-2-expressing cells in vitro. A single dose of VEGFR-2 CAR-engineered mouse T cells plus exogenous IL-2 significantly inhibited the growth of 5 different types of established, vascularized syngeneic tumors in 2 different strains of mice and prolonged the survival of mice. T cells transduced with VEGFR-2 CAR showed durable and increased tumor infiltration, correlating with their antitumor effect. This approach provides a potential method for the gene therapy of a variety of human cancers.
Figures


References
-
- Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6(4):389–395. - PubMed
-
- Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6(4):273–286. - PubMed
-
- Padro T, et al. Overexpression of vascular endothelial growth factor (VEGF) and its cellular receptor KDR (VEGFR-2) in the bone marrow of patients with acute myeloid leukemia. Leukemia. 2002;16(7):1302–1310. - PubMed
