

Mechanisms of drug toxicity and relevance to pharmaceutical development
- PMID: 20978361
- PMCID: PMC4707670
- DOI: 10.2133/dmpk.dmpk-10-rv-062
Mechanisms of drug toxicity and relevance to pharmaceutical development
Abstract
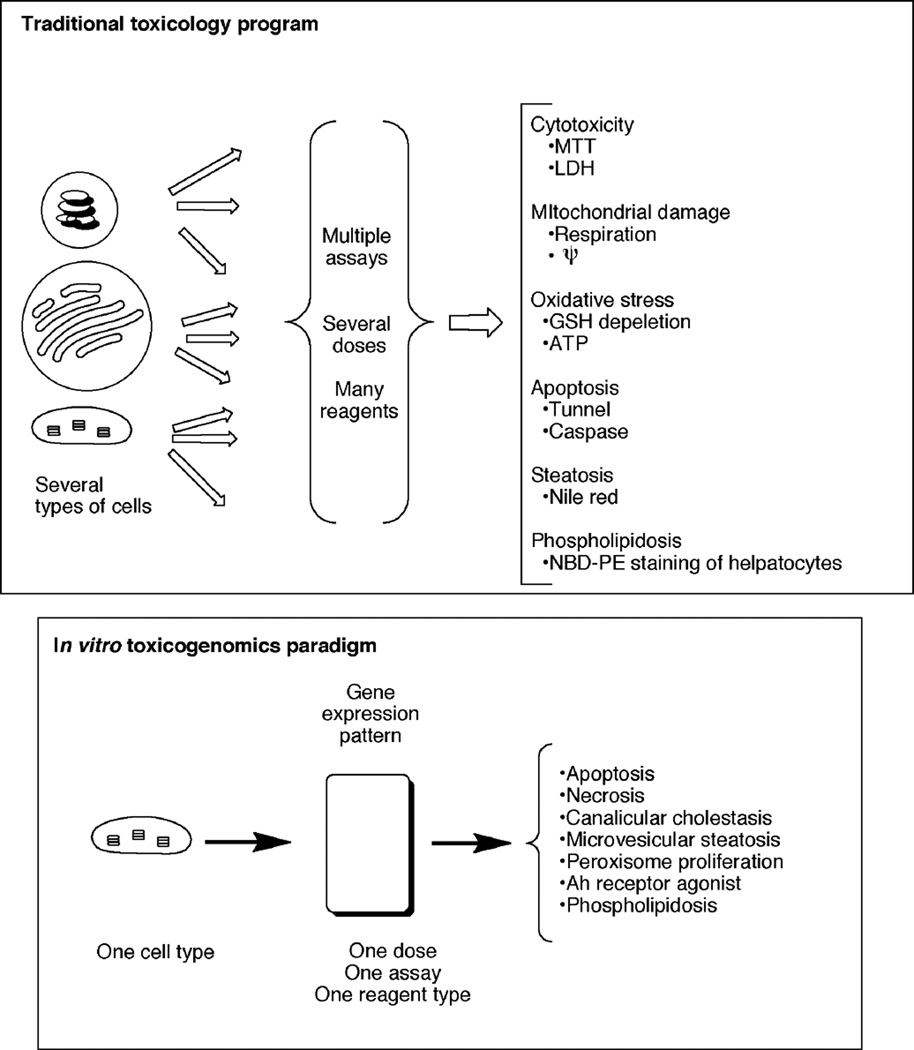
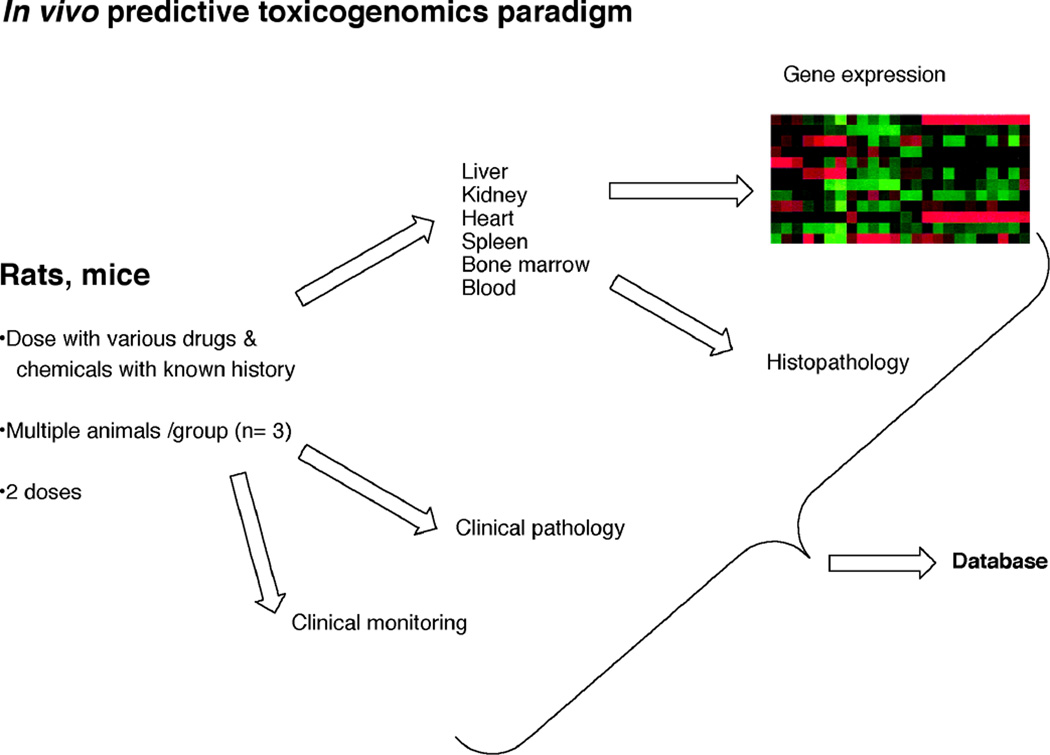
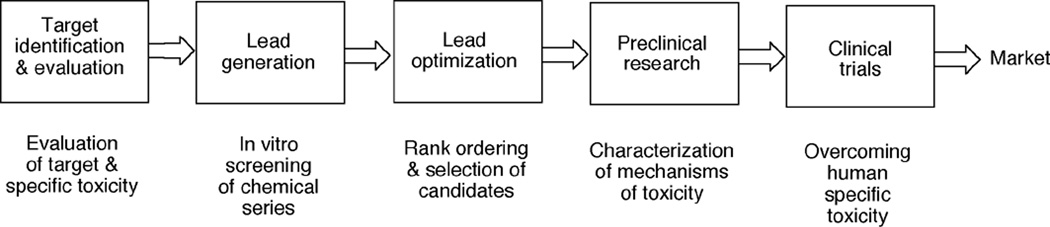
Toxicity has been estimated to be responsible for the attrition of approximately one-third of drug candidates and is a major contributor to the high cost of drug development, particularly when not recognized until late in clinical trials or post-marketing. The causes of drug toxicity can be classified in several ways and include mechanism-based (on-target) toxicity, immune hypersensitivity, off-target toxicity, and bioactivation/covalent modification. In addition, idiosyncratic responses are rare but can be one of the most problematic issues; several hypotheses for these have been advanced. Although covalent binding of drugs to proteins was described almost 40 years ago, the significance to toxicity has been difficult to establish; recent literature in this field is considered. The development of more useful biomarkers and short-term assays for rapid screening of drug toxicity early in the drug discovery/development process is a major goal, and some progress has been made using "omics" approaches.
Figures
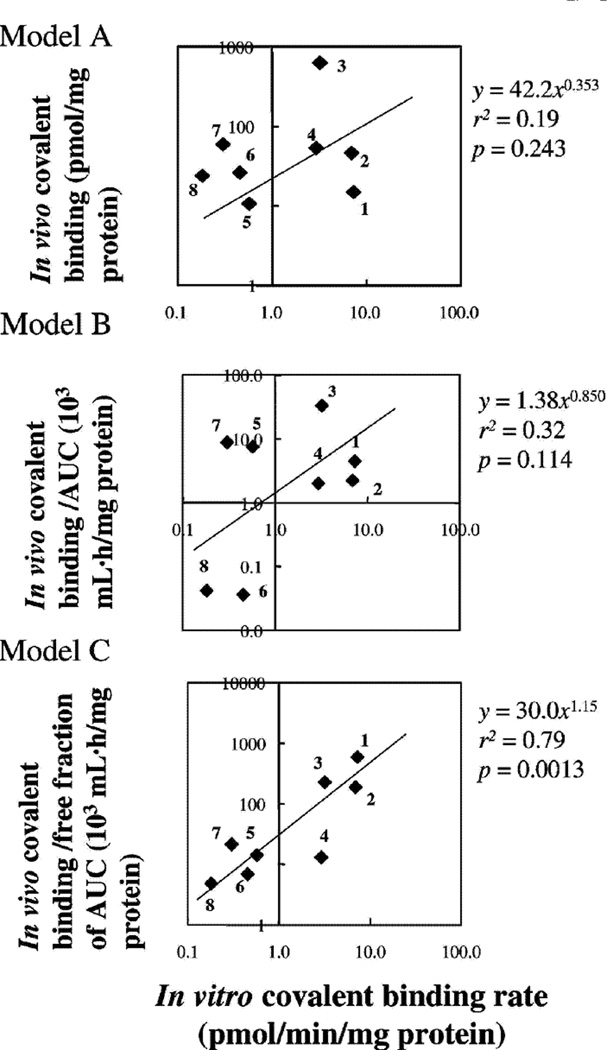
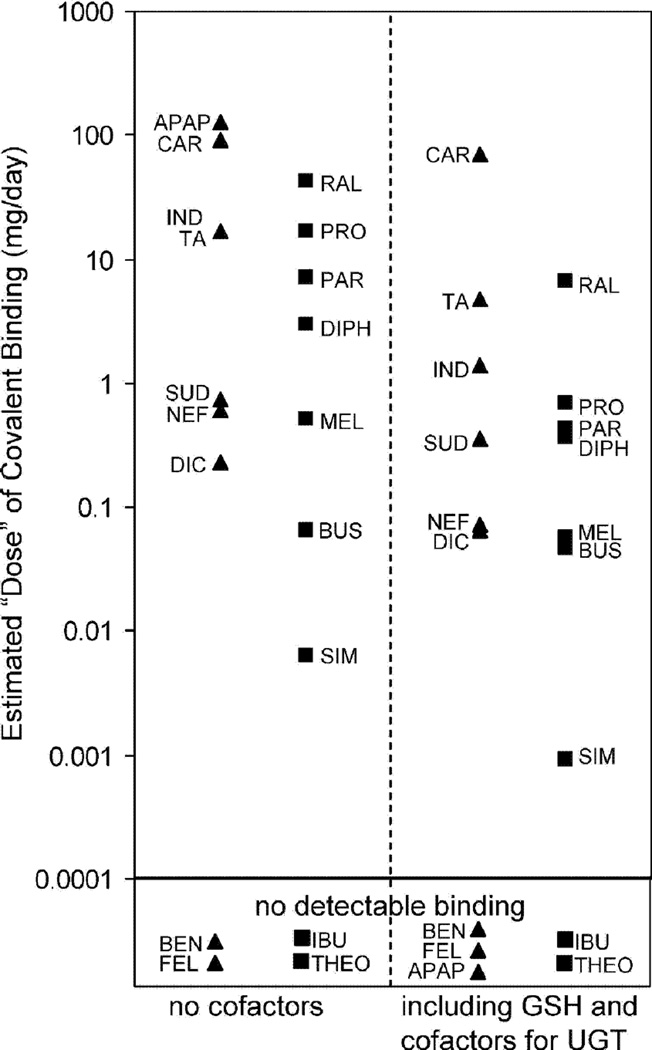
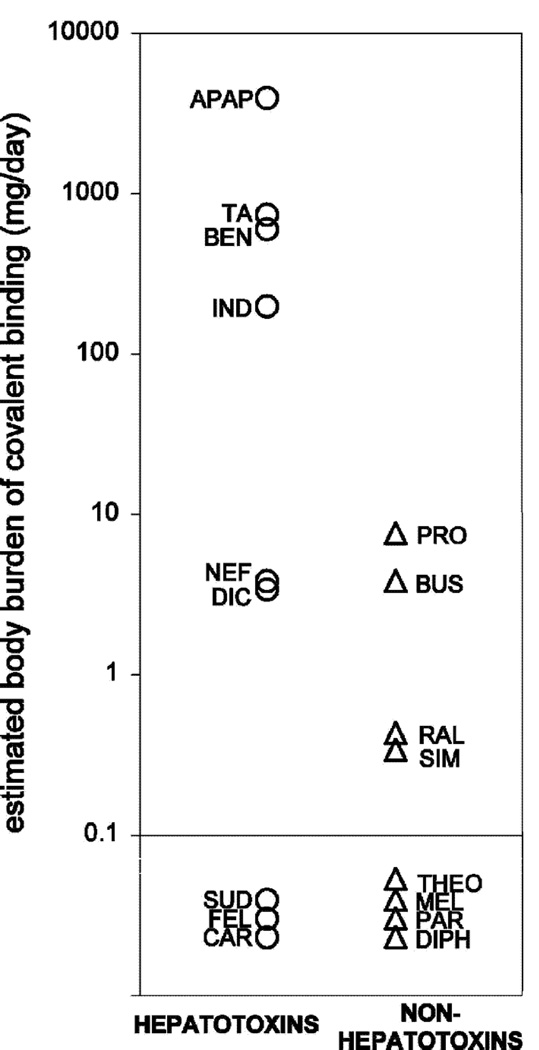
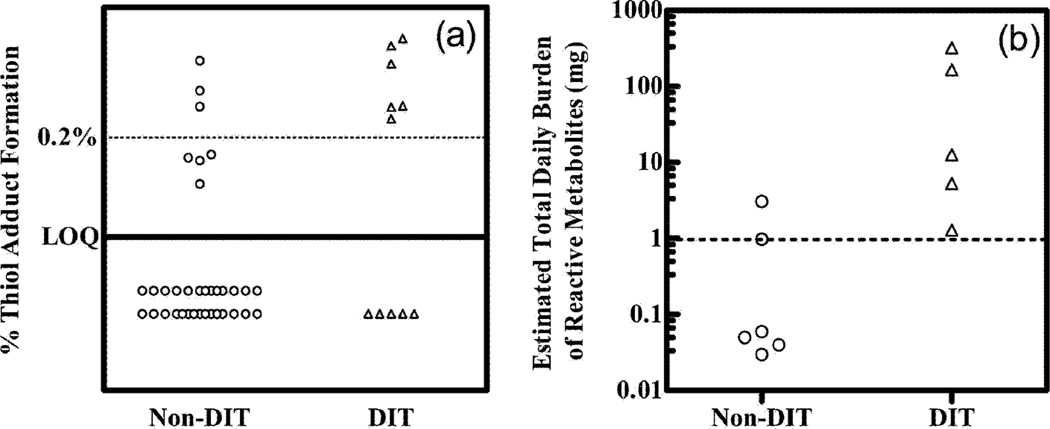
References
-
- U. S. Food and Drug Administration. Challenge and opportunity on the critical path to new medical products. US Department of Health and Human Services; 2004. Mar,
-
- Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004;3:711–715. - PubMed
-
- Borzelleca JF. Profiles in Toxicology - Paracelsus: herald of modern toxicology. Toxicol. Sci. 2000;53:2–4. - PubMed
-
- Liebler DC, Guengerich FP. Elucidating mechanisms of drug-induced toxicity. Nat. Rev. Drug Discov. 2005;4:410–420. - PubMed
-
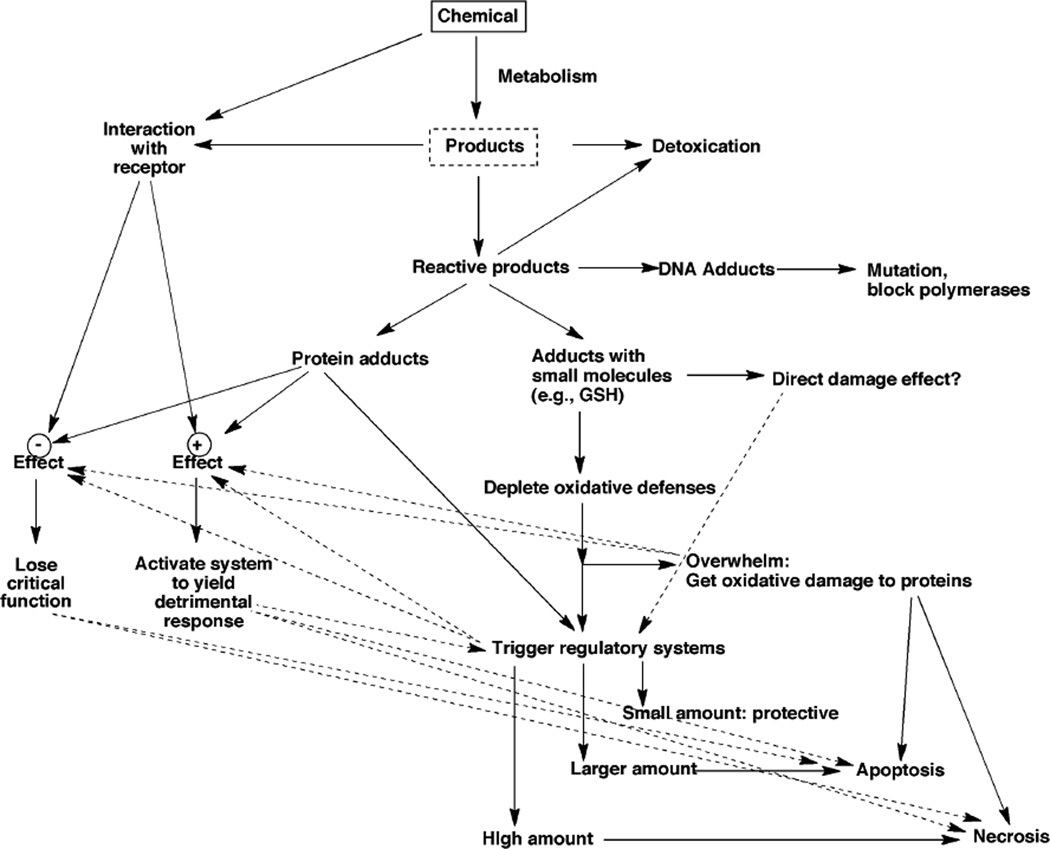
- Park BK, Kitteringham NR, Maggs JL, Pirmohamed M, Williams DP. The role of metabolic activation in drug-induced hepatotoxicity. Annu. Rev. Pharmacol. Toxicol. 2005;45:177–202. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical