

Mitochondrial cardiomyopathies: how to identify candidate pathogenic mutations by mitochondrial DNA sequencing, MITOMASTER and phylogeny
- PMID: 20978534
- PMCID: PMC3025796
- DOI: 10.1038/ejhg.2010.169
Mitochondrial cardiomyopathies: how to identify candidate pathogenic mutations by mitochondrial DNA sequencing, MITOMASTER and phylogeny
Abstract
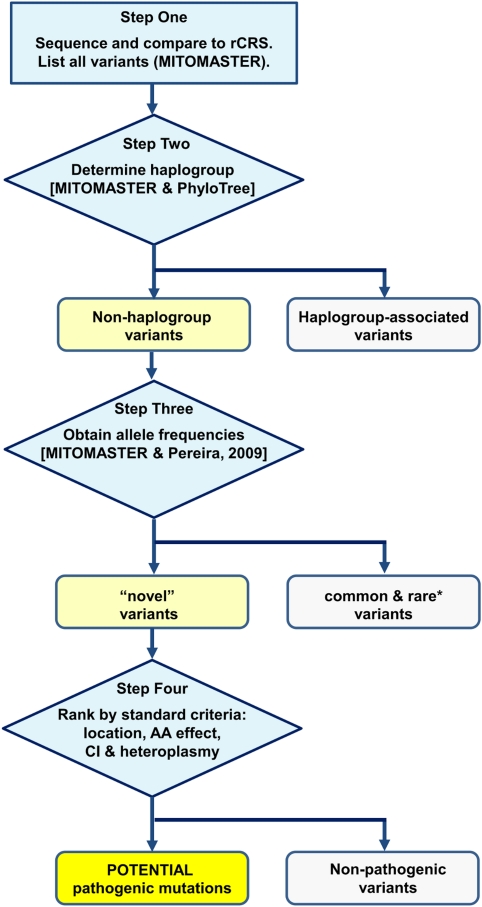
Pathogenic mitochondrial DNA (mtDNA) mutations leading to mitochondrial dysfunction can cause cardiomyopathy and heart failure. Owing to a high mutation rate, mtDNA defects may occur at any nucleotide in its 16 569 bp sequence. Complete mtDNA sequencing may detect pathogenic mutations, which can be difficult to interpret because of normal ethnic/geographic-associated haplogroup variation. Our goal is to show how to identify candidate mtDNA mutations by sorting out polymorphisms using readily available online tools. The purpose of this approach is to help investigators in prioritizing mtDNA variants for functional analysis to establish pathogenicity. We analyzed complete mtDNA sequences from 29 Italian patients with mitochondrial cardiomyopathy or suspected disease. Using MITOMASTER and PhyloTree, we characterized 593 substitution variants by haplogroup and allele frequencies to identify all novel, non-haplogroup-associated variants. MITOMASTER permitted determination of each variant's location, amino acid change and evolutionary conservation. We found that 98% of variants were common or rare, haplogroup-associated variants, and thus unlikely to be primary cause in 80% of cases. Six variants were novel, non-haplogroup variants and thus possible contributors to disease etiology. Two with the greatest pathogenic potential were heteroplasmic, nonsynonymous variants: m.15132T>C in MT-CYB for a patient with hypertrophic dilated cardiomyopathy and m.6570G>T in MT-CO1 for a patient with myopathy. In summary, we have used our automated information system, MITOMASTER, to make a preliminary distinction between normal mtDNA variation and pathogenic mutations in patient samples; this fast and easy approach allowed us to select the variants for traditional analysis to establish pathogenicity.
Figures


References
-
- Wallace DC, Brown MD, Lott MT. Mitochondrial DNA variation in human evolution and disease. Gene. 1999;238:211–230. - PubMed
-
- Marin-Garcia J, Goldenthal MJ. Cardiomyopathy and abnormal mitochondrial function. Cardiovasc Res. 1994;28:456–463. - PubMed
-
- Anderson S, Bankier AT, Barrell BG, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. - PubMed
-
- Brega A, Narula J, Arbustini E. Functional, structural, and genetic mitochondrial abnormalities in myocardial diseases. J Nucl Cardiol. 2001;8:89–97. - PubMed
