

Adenovirus membrane penetration activates the NLRP3 inflammasome
- PMID: 20980503
- PMCID: PMC3014182
- DOI: 10.1128/JVI.01265-10
Adenovirus membrane penetration activates the NLRP3 inflammasome
Abstract
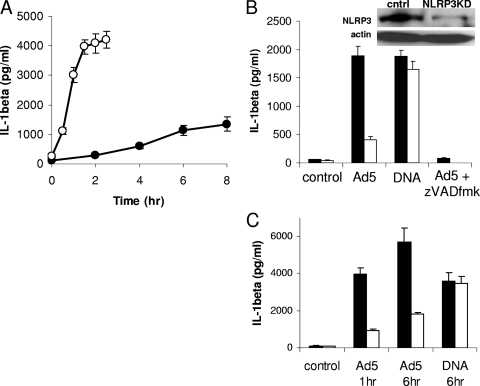
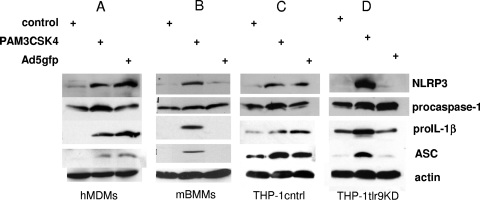
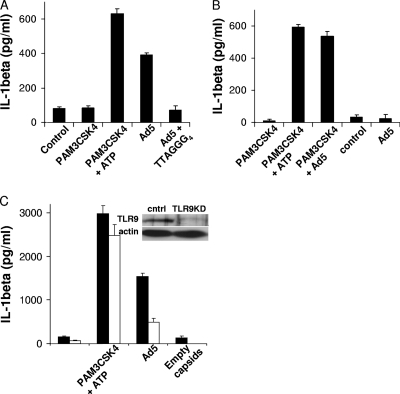
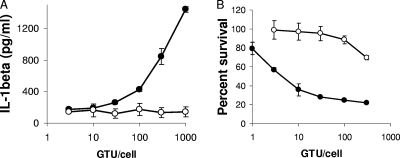
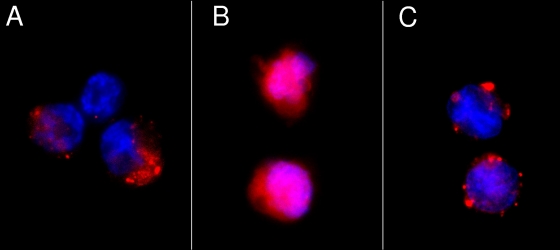
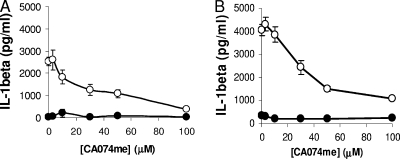
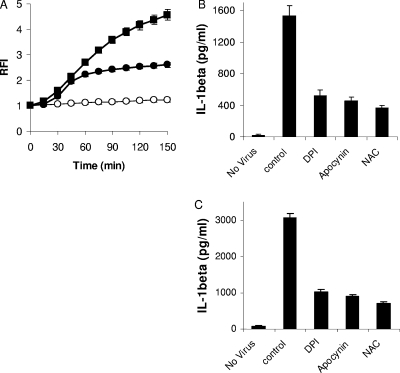
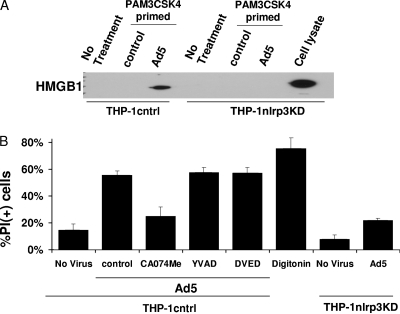
Adenovirus type 5 (Ad5) infection of macrophages results in rapid secretion of interleukin-1β (IL-1β) and is dependent on the inflammasome components NLRP3 and ASC and the catalytic activity of caspase-1. Using lentivirus-expressed short hairpin RNA (shRNA) and competitive inhibitors, we show that Ad-induced IL-1β release is dependent upon Toll-like receptor 9 (TLR9) sensing of the Ad5 double-stranded DNA (dsDNA) genome in human cell lines and primary monocyte-derived macrophages but not in mouse macrophages. Additionally, a temperature-sensitive mutant of Ad5 unable to penetrate endosomal membranes, ts1, is unable to induce IL-1β release in TLR2-primed THP-1 cells, suggesting that penetration of endosomal membranes is required for IL-1β release. Disruption of lysosomal membranes and the release of cathepsin B into the cytoplasm are required for Ad-induced NLRP3 activation. Ad5 cell entry also induces reactive oxygen species (ROS) production, and inhibitors of ROS prevent Ad-induced IL-1β release. Ad5 activation of NLRP3 also induces necrotic cell death, resulting in the release of the proinflammatory molecule HMGB1. This work further defines the mechanisms of virally induced inflammasome activation.
Figures
References
-
- Anonymous. 2007. Acute respiratory disease associated with adenovirus serotype 14—four states, 2006-2007. MMWR Morb. Mortal. Wkly. Rep. 56:1181-1184. - PubMed
-
- Appledorn, D. M., S. Patial, A. McBride, S. Godbehere, N. Van Rooijen, N. Parameswaran, and A. Amalfitano. 2008. Adenovirus vector-induced innate inflammatory mediators, MAPK signaling, as well as adaptive immune responses are dependent upon both TLR2 and TLR9 in vivo. J. Immunol. 181:2134-2144. - PubMed
-
- Bauernfeind, F. G., G. Horvath, A. Stutz, E. S. Alnemri, K. Macdonald, D. Speert, T. Fernandes-Alnemri, J. Wu, B. G. Monks, K. A. Fitzgerald, V. Hornung, and E. Latz. 2009. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183:787-791. - PMC - PubMed
-
- Benko, M., and B. Harrach. 2003. Molecular evolution of adenoviruses. Curr. Top. Microbiol. Immunol. 272:3-35. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
